01 研究背景
心臟重構(gòu)是各種心血管疾病發(fā)展過(guò)程中常見(jiàn)的病理生理過(guò)程,但目前仍缺乏有效的干預(yù)措施。腫瘤壞死受體相關(guān)因子7(TRAF7)屬于腫瘤壞死因子受體相關(guān)因子家族,在生物過(guò)程中起重要作用。先前的研究表明,TRAF7突變會(huì)導(dǎo)致先天性心臟缺陷和畸形。然而,TRAF7在病理性心肌肥厚發(fā)病機(jī)制中的分子機(jī)制尚不清楚。武漢大學(xué)人民醫(yī)院唐其柱教授團(tuán)隊(duì)在Cardiovascular Research (IF 10.2)上發(fā)表文章“Cardiac tumour necrosis factor receptor-associated factor 7 mediates the ubiquitination of apoptosis signal-regulating kinase 1 and aggravates cardiac hypertrophy”,發(fā)現(xiàn)TRAF7是心臟肥厚過(guò)程中一個(gè)重要的調(diào)節(jié)因子,TRAF7直接與凋亡信號(hào)調(diào)節(jié)激酶-1(ASK1)相互作用,并在PE刺激下通過(guò)介導(dǎo)ASK1的K63連鎖泛素化來(lái)促進(jìn)ASK1磷酸化,進(jìn)而促進(jìn)心臟肥大過(guò)程中ASK1的激活和下游信號(hào)傳導(dǎo),表明調(diào)控TRAF7和ASK1可能是一種新的心肌肥厚治療策略。

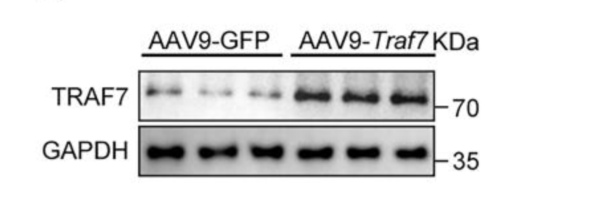
AAV9-cTNT-Traf7在小鼠心臟中過(guò)表達(dá)TRAF7

02 結(jié)果展示
1. 心臟特異性的TRAF7過(guò)表達(dá)加速心臟重構(gòu)
作者發(fā)現(xiàn)TRAF7在實(shí)驗(yàn)性肥厚模型中表達(dá)升高,并能在體外促進(jìn)心肌細(xì)胞肥大。為研究TRAF7的作用機(jī)制,作者用AAV9-cTNT-Traf7在小鼠心臟中過(guò)表達(dá)TRAF7,發(fā)現(xiàn)TRAF7的過(guò)表達(dá)加重了TAC誘導(dǎo)的心肌肥厚、心臟重量增加,惡化了左室收縮功能,HW/BW、LW/BW、HW/TL比值均增加。HE染色顯示TAC4周后,TRAF7過(guò)表達(dá)小鼠的心肌細(xì)胞CSA顯著增加。PSR檢測(cè)顯示TAC誘導(dǎo)的心臟間質(zhì)纖維化和血管周?chē)w維化均因TRAF7過(guò)表達(dá)而加重。此外TRAF7過(guò)表達(dá)組心肌肥厚和心肌纖維化相關(guān)基因mRNA水平均顯著升高。這些結(jié)果表明TRAF7過(guò)表達(dá)加速心功能障礙、心臟肥厚和心臟重構(gòu)。進(jìn)一步研究發(fā)現(xiàn)TRAF7通過(guò)促進(jìn)ASK1的磷酸化來(lái)調(diào)節(jié)ASK1-JNK1/2/p38軸,進(jìn)而促進(jìn)心臟肥厚。
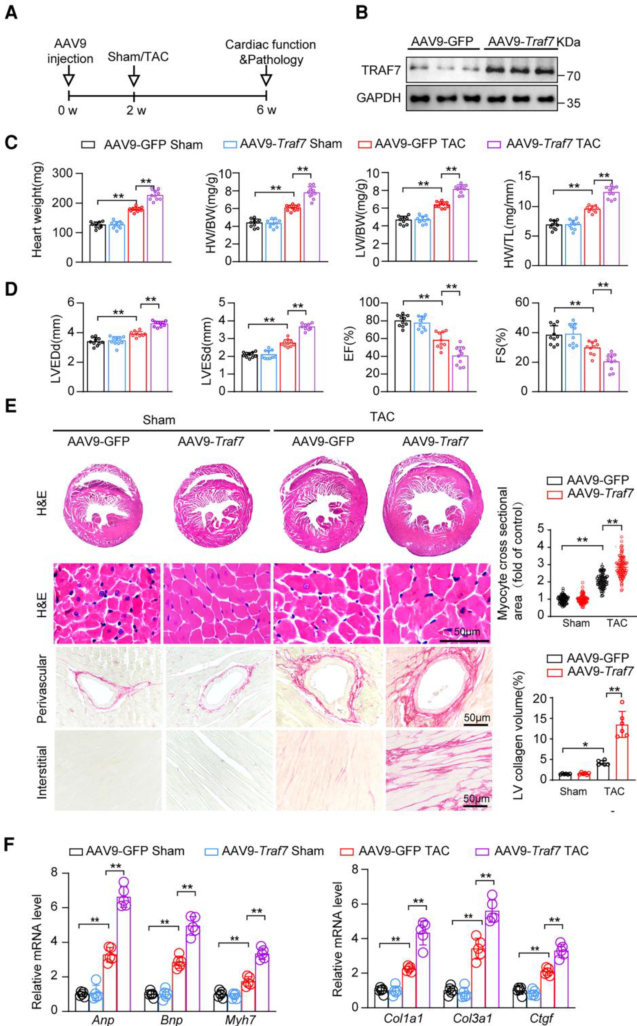
圖1 心臟特異性的TRAF7過(guò)表達(dá)加速心臟重構(gòu)
2. TRAF7通過(guò)促進(jìn)ASK1泛素化調(diào)節(jié)ASK1的激活
作者通過(guò)IP、GST下拉、雙免疫熒光染色和Co-IP等實(shí)驗(yàn)結(jié)果證明ASK1與TRAF7存在相互作用。由于TRAF7具有E3泛素連接酶活性,接下來(lái)作者研究了TRAF7是否通過(guò)其E3泛素連接酶活性影響ASK1的活性。研究結(jié)果表明PE明顯觸發(fā)了NRCM(新生大鼠心肌細(xì)胞)中ASK1泛素化,并提高了ASK1磷酸化水平,假手術(shù)或TAC手術(shù)的野生型(WT)和AAV9-Traf7小鼠的體內(nèi)泛素化實(shí)驗(yàn)中也檢測(cè)了ASK1的泛素化和磷酸化,TRAF7過(guò)表達(dá)上調(diào)ASK1的泛素化和磷酸化。此外,在過(guò)表達(dá)TRAF7的293T細(xì)胞中也觀察到ASK1泛素化和磷酸化的增加,表明ASK1泛素化和磷酸化是相關(guān)的,而TRAF7可以促進(jìn)這兩個(gè)過(guò)程。進(jìn)一步研究發(fā)現(xiàn)過(guò)表達(dá)TRAF7導(dǎo)致ASK1的k63連鎖泛素化水平升高,K1064是負(fù)責(zé)TRAF7介導(dǎo)的Lys63連鎖泛素化的泛素化位點(diǎn),從而觸發(fā)ASK1磷酸化活化。
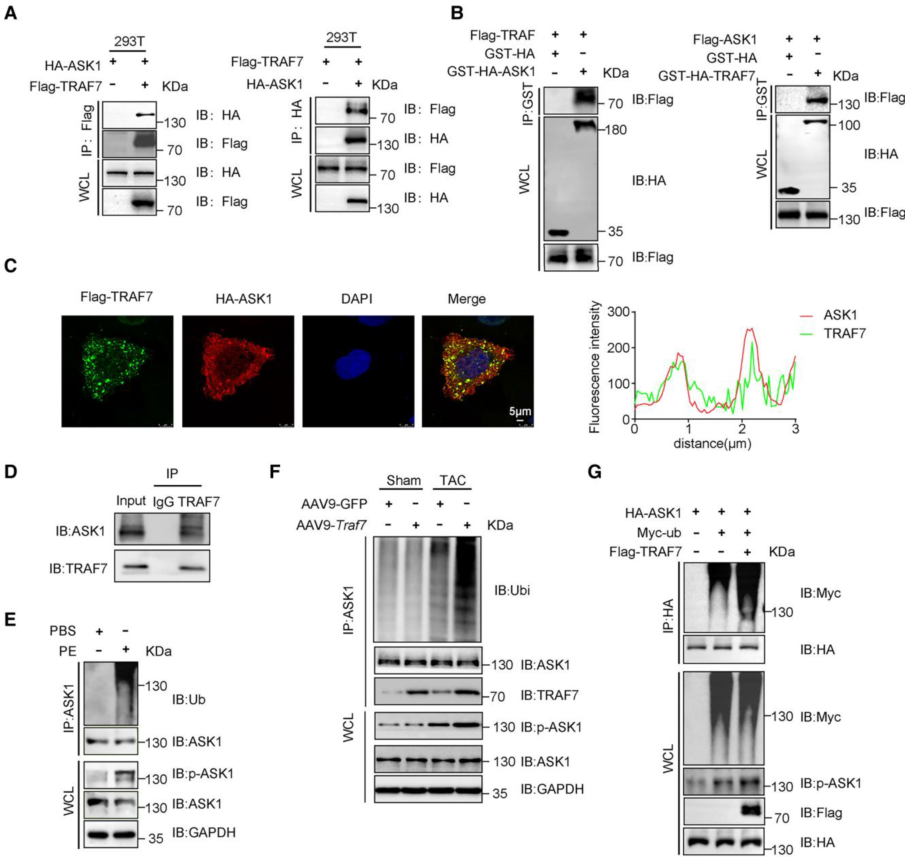
圖2 TRAF7通過(guò)促進(jìn)ASK1泛素化調(diào)節(jié)ASK1的激活
3. ASK1是TRAF7誘導(dǎo)心肌肥厚的靶點(diǎn)
為了進(jìn)一步探討TRAF7是否通過(guò)激活A(yù)SK1調(diào)控心肌肥大,作者在TRAF7過(guò)表達(dá)的情況下,利用ASK1特異性抑制劑GS4997抑制ASK1激活。結(jié)果顯示,與PE刺激后TRAF7過(guò)表達(dá)相比,GS4997挽救了細(xì)胞的CSA,降低了TRAF7過(guò)表達(dá)上調(diào)的Anp、Bnp和Myh7 mRNA水平。此外TRAF7誘導(dǎo)的PE刺激后ASK1/JNK/p38軸的激活被GS4997抑制。然后作者用Ask1-CKO小鼠在體內(nèi)進(jìn)行了拯救實(shí)驗(yàn),結(jié)果顯示,Ask1-CKO能夠顯著拯救壓力過(guò)載誘導(dǎo)的心臟重構(gòu),同時(shí)也能夠抑制TRAF7過(guò)表達(dá)誘導(dǎo)的嚴(yán)重心臟重構(gòu)。這表明在TAC誘導(dǎo)的心肌肥厚過(guò)程中,ASK1是TRAF7的關(guān)鍵下游靶點(diǎn)。
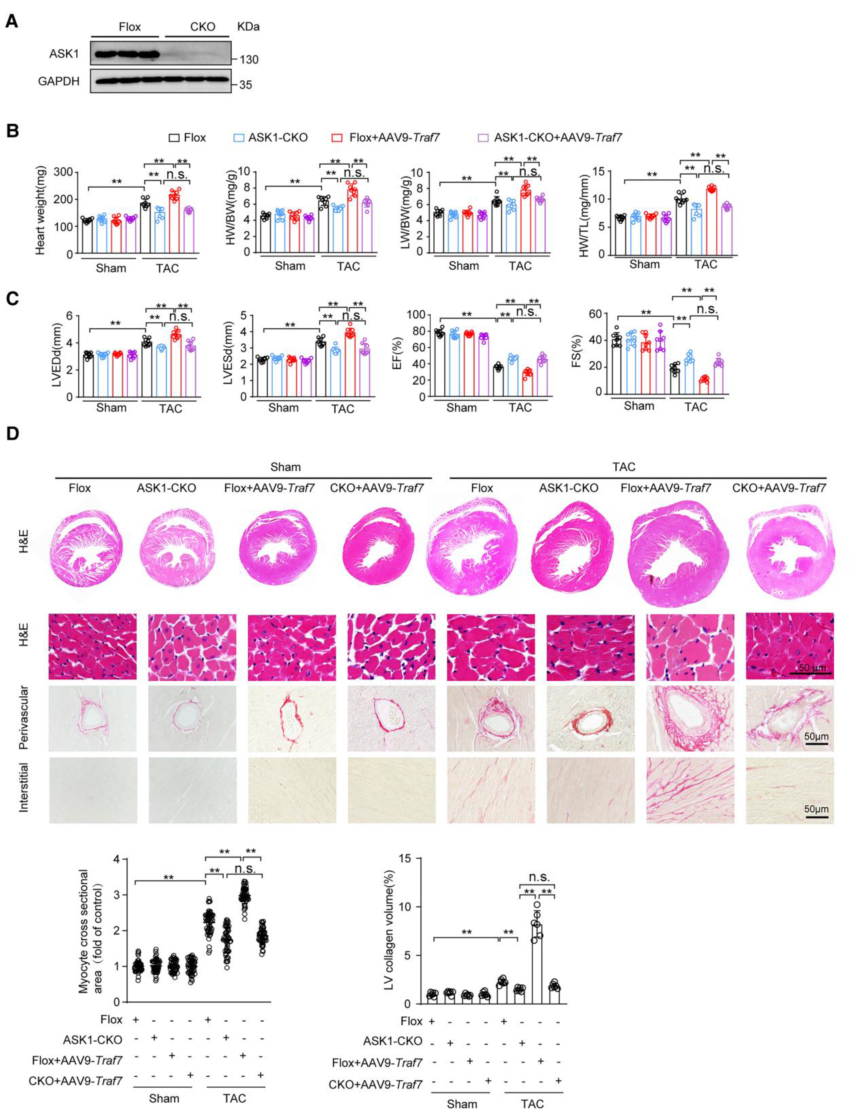
圖3 ASK1是TRAF7誘導(dǎo)心肌肥厚的靶點(diǎn)
03 實(shí)驗(yàn)結(jié)論
本研究發(fā)現(xiàn)TRAF7在心臟肥厚過(guò)程中是一個(gè)重要的調(diào)節(jié)因子,TRAF7直接與ASK1相互作用,并在PE刺激下通過(guò)介導(dǎo)ASK1的K63連鎖泛素化來(lái)促進(jìn)ASK1磷酸化,進(jìn)而促進(jìn)心臟肥大過(guò)程中ASK1的激活和下游信號(hào)傳導(dǎo),調(diào)控TRAF7和ASK1可能是一種新的心肌肥厚治療策略。