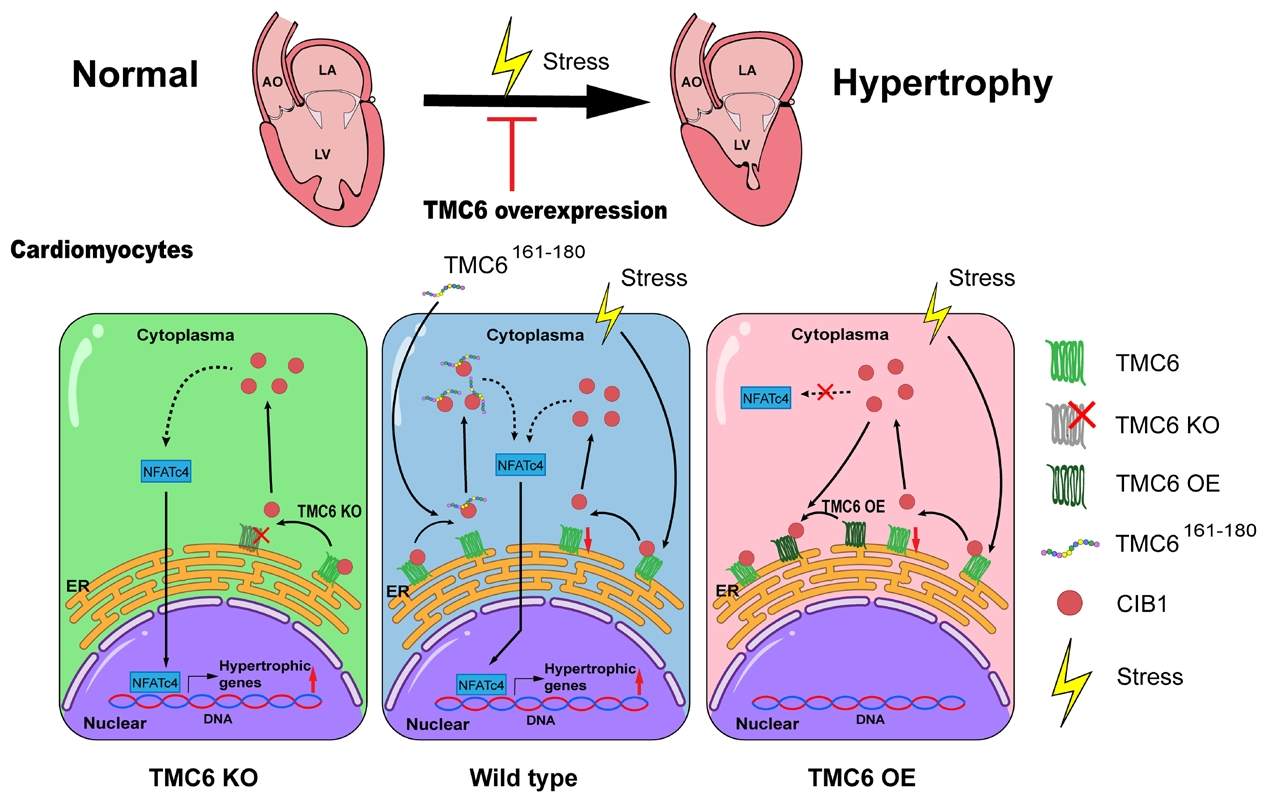
心肌肥厚是心力衰竭的主要原因之一,但有效的治療靶點仍然有限,而跨膜通道樣蛋白6(TMC6)在健康心肌中含量豐富,但在肥厚心肌中表達下調,其在心肌肥厚中的作用尚不清楚。浙江大學醫學院附屬第一醫院梁平教授團隊聯合復旦大學腦科學研究院唐逸泉教授團隊在Circulation Research(IF 16.2)發表題為“TMC6 Is a Novel Therapeutic Target for Pathogenic Cardiac Hypertrophy”的文章。研究揭示TMC6是病理性心肌肥厚的內源性抑制因子,通過將CIB1隔離在內質網中,從而抑制CIB1-鈣調神經磷酸酶/NFAT信號通路,提示TMC6–CIB1軸是一個潛在的治療靶點。

· 維真助力·
基因信息 TMC6:跨膜通道樣蛋白6
實驗動物 8周齡WT和Tmc6敲除小鼠
病毒產品 AAV9-cTNT-TMC6;AAV9-cTNT-GFP
注射方式 尾靜脈注射
病毒用量 1×1012 genomic copies/mouse
研究結果
1、TMC6缺失加重心肌肥厚,過表達則起保護作用
研究數據顯示在人類與小鼠心臟中,TMC6是TMC家族中表達最高的成員,并且在三種心肌肥厚模型中,TMC6蛋白水平均顯著下降。心肌特異性Tmc6敲除小鼠出現輕度肥厚,并在TAC后表現出更嚴重的心功能下降、肥大、纖維化和生存率降低。為了進一步研究TMC6是否可以以細胞自主的方式預防壓力超負荷誘導的心臟肥大,研究團隊將AAV9-cTNT-TMC6及對照病毒注射至WT和Tmc6敲除小鼠體內;載體給藥后4周,小鼠接受TAC手術。與表達GFP的對照組相比,心臟特異性過表達TMC6的WT-TAC和KO-TAC小鼠顯著降低了心臟肥大和功能障礙,體現TMC6在防止TAC誘導的心肌肥大中的細胞自主作用。進一步的RNA-Seq分析表明Tmc6基因敲除通過激活特定的TF,如SRF,MEF家族和NFAT家族,加劇心肌肥厚。
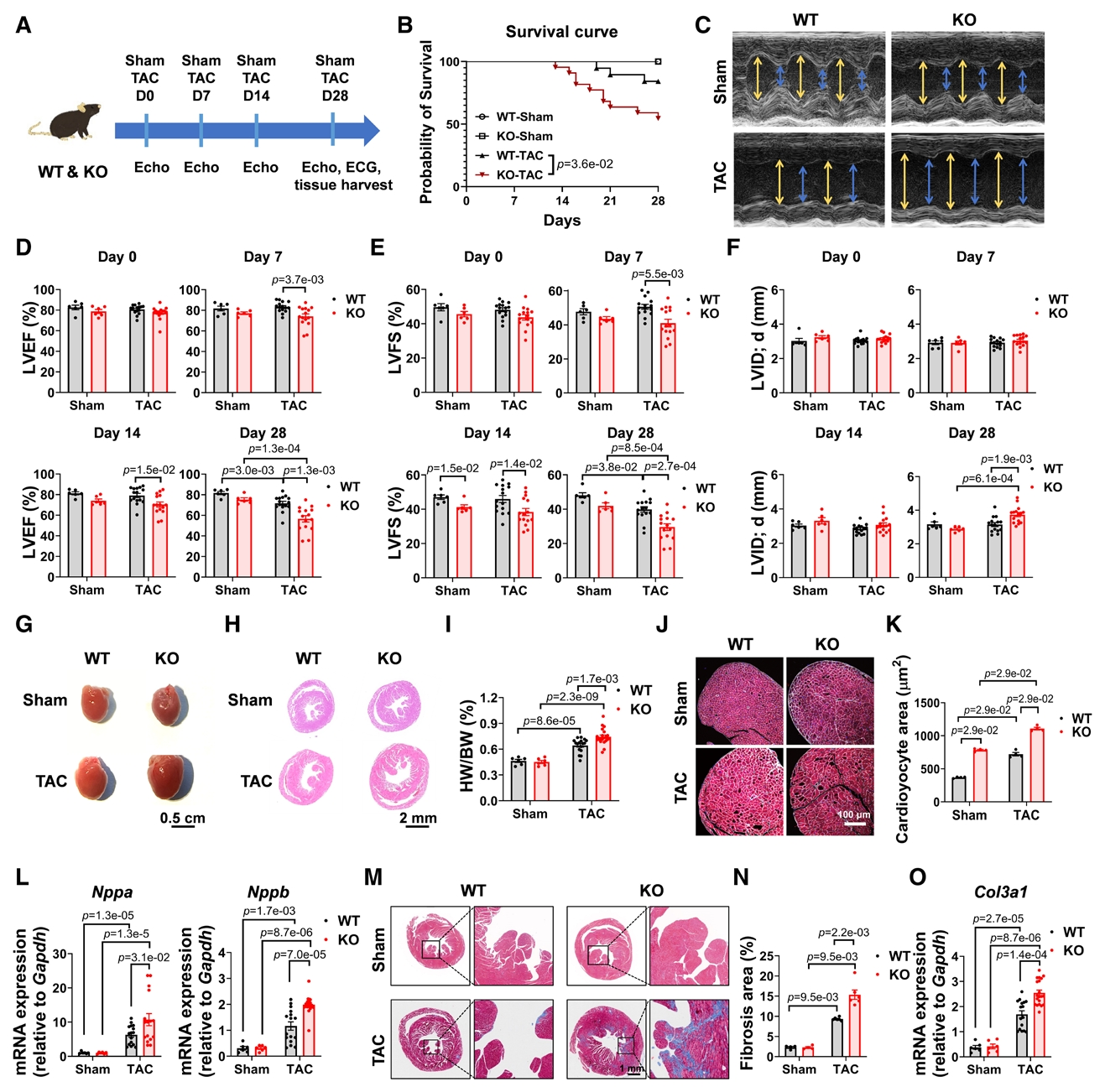
TMC6缺陷加重小鼠壓力超負荷后的心臟肥大
2、TMC6與CIB1結合以抑制CIB1依賴性心臟肥大
體外TMC6敲低及敲除的細胞在肥厚刺激下,鈣調神經磷酸酶活性升高、NFATc4 核轉位增加,肥厚標志物表達上調,該效應可被鈣調神經磷酸酶抑制劑逆轉,表明TMC6缺乏可通過激活鈣調磷酸酶/NFAT信號通路導致心肌細胞肥大。通過酵母雙雜交篩選及免疫共沉淀等分析,證實TMC6與CIB1存在相互作用,TMC6的161-180氨基酸片段為CIB1結合域,該片段可競爭性破壞TMC6-CIB1結合。亞細胞定位實驗發現TMC6定位于內質網(ER),當CIB1單獨表達時,主要定位于質膜和細胞核,在與TMC6共表達時,CIB1則重新定位于ER;此外競爭性肽TMC6161-180的共表達可阻斷這種重新定位。實驗進一步證實敲除CIB1能夠逆轉因TMC6缺失所誘發的心肌肥厚表型;TMC6161-180將CIB1從TMC6上置換下來,從而增加了游離CIB1的池容量,使其能夠在肌膜微區招募鈣調神經磷酸酶并激活NFAT依賴的促肥厚信號通路。
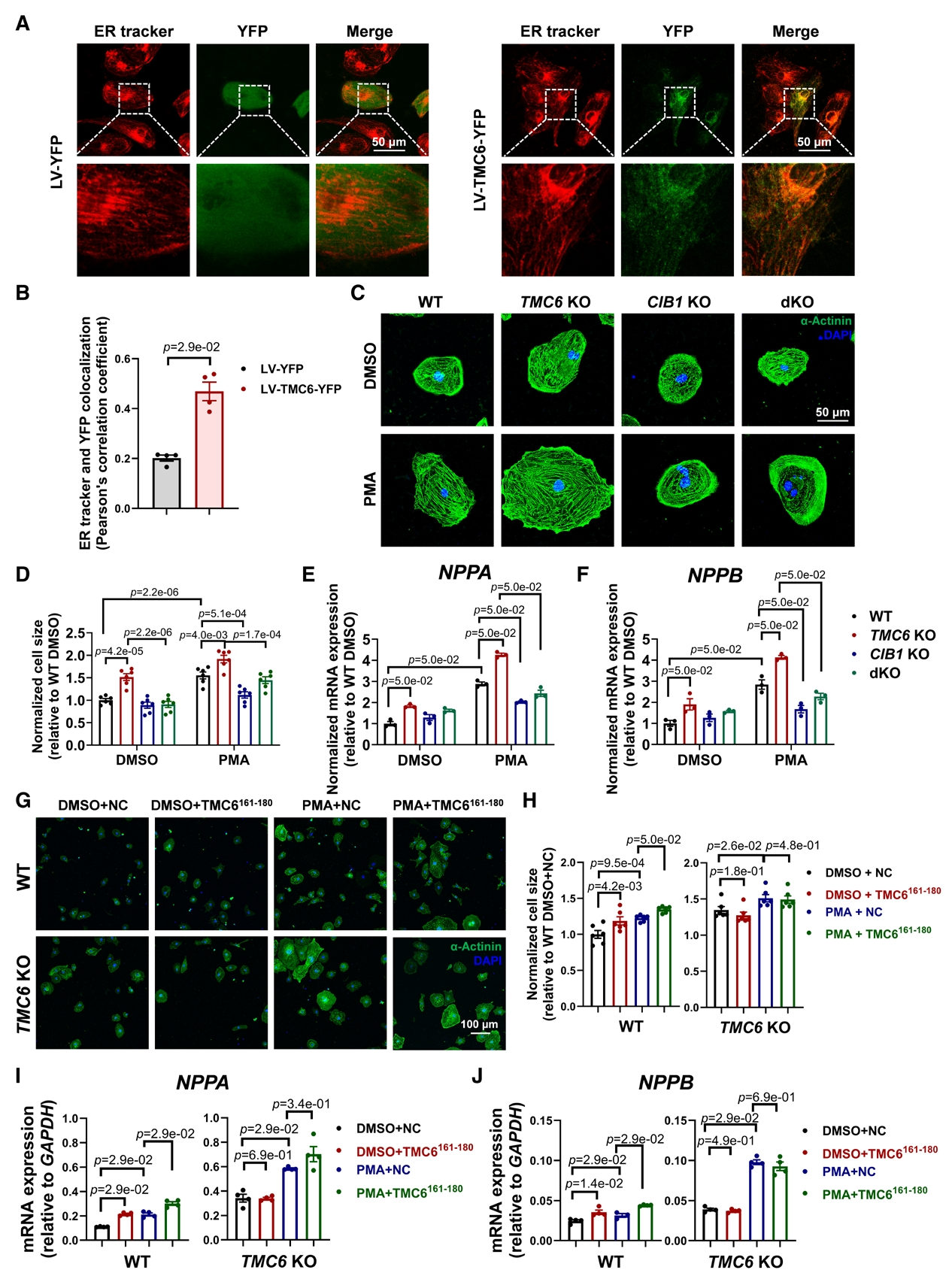
TMC6與CIB1結合,抑制CIB1依賴性心臟肥大
3、TMC6基因治療病理性心肌肥厚的研究進展
基于上述的結果,研究團隊提出假設:TMC6可能是心肌肥厚的一個治療靶點,隨之進行了體外和體內研究。體外過表達TMC6可下調PMA誘導的肥大基因表達,并使細胞體積縮小。為了研究TMC6基因治療對體內心臟肥大的治療效果,WT小鼠在假手術或TAC手術后4周接受尾靜脈注射AAV9-cTNT-TMC6或AAV9-cTNT-GFP。評估結果顯示TMC6過表達顯著恢復了TMC6蛋白水平,改善了心臟功能,減輕了心肌細胞的肥大以及心臟纖維化。這些結果表明,TMC6基因治療有效地減輕體外和體內模型的病理性心臟肥大。
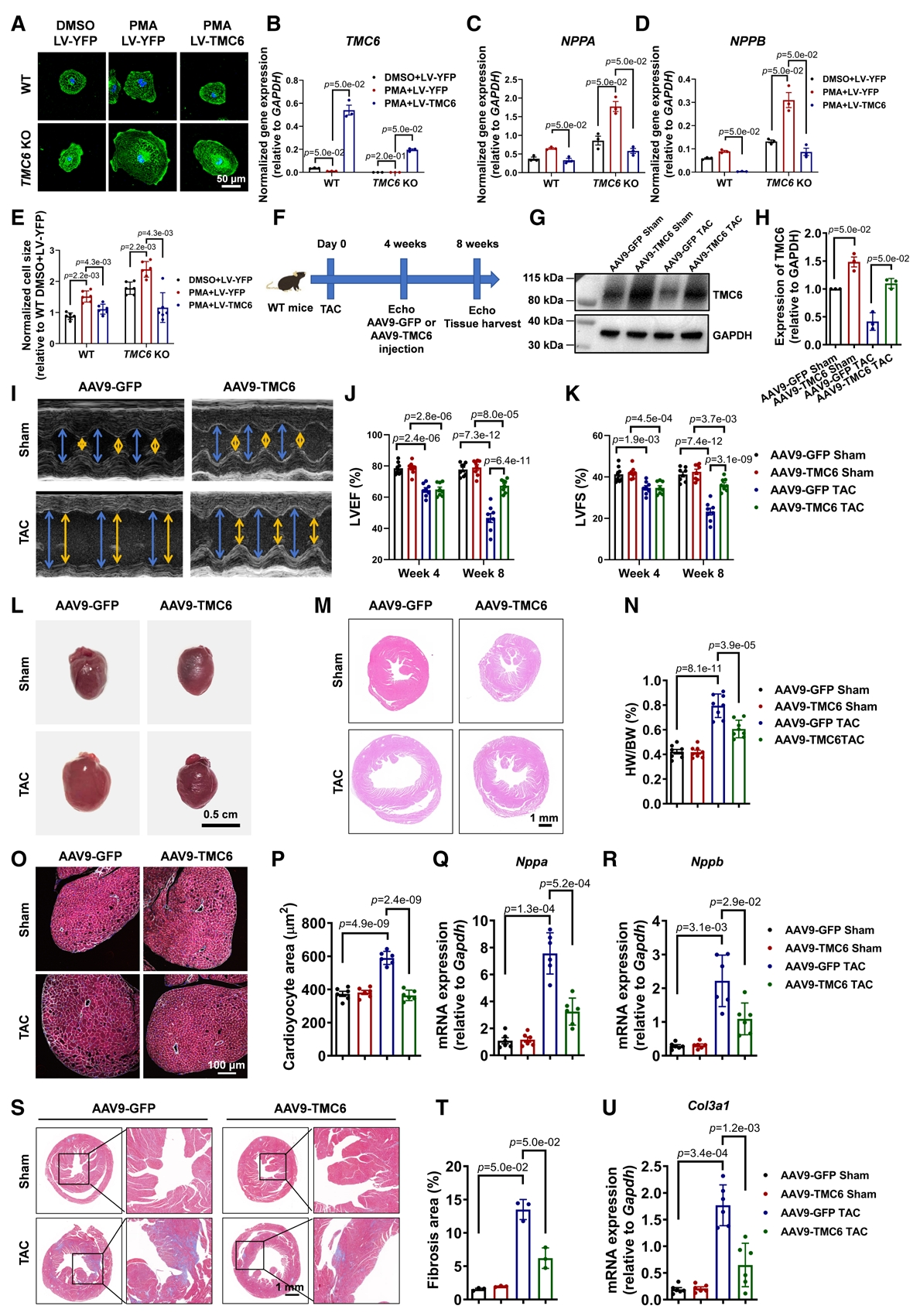
TMC6基因治療病理性心肌肥厚的研究
研究結論
研究揭示了TMC6在心肌肥厚中的新作用,證明其通過CIB1-鈣調神經磷酸酶-NFAT信號通路發揮抗肥厚效應。這些發現表明,TMC6是治療病理性心肌肥厚和心力衰竭的潛在治療靶點。