癌癥蛋白質(zhì)基因組學(xué)涵蓋了基因組學(xué)、表觀基因組學(xué)、轉(zhuǎn)錄組學(xué)、蛋白質(zhì)組學(xué)和蛋白質(zhì)修飾組學(xué)等多個維度的數(shù)據(jù)信息。基因組學(xué)和表觀遺傳學(xué)提供我們的是將要發(fā)生什么的細(xì)胞藍(lán)圖,而蛋白質(zhì)組學(xué)及蛋白質(zhì)修飾組學(xué)告訴我們的是已經(jīng)發(fā)生的確定事件。蛋白質(zhì)基因組學(xué)有助于解析癌癥的發(fā)生和發(fā)展機(jī)制,為癌癥的精準(zhǔn)分型與個體化治療、療效監(jiān)測和預(yù)后判斷提供了新的思路和策略。
近日,Broad研究所等團(tuán)隊(duì)在Nature Reviews Cancer(IF=60.716)發(fā)表了題為"Cancer proteogenomics: current impact and future prospects"的重磅綜述文章,對蛋白質(zhì)基因組學(xué)在腫瘤研究中的最新進(jìn)展進(jìn)行了系統(tǒng)總結(jié),并對蛋白質(zhì)基因組學(xué)中樣本收集、分析方法、臨床應(yīng)用等展開了分析討論。

01.癌癥蛋白質(zhì)組學(xué)概覽
癌癥基因組計(jì)劃(Cancer Genome Project)執(zhí)行以來,人類對于自身基因組的了解日益深入,癌癥的靶向治療手段逐漸發(fā)展,然而絕大多數(shù)體細(xì)胞突變屬于沒有特定致癌功能的乘客突變(passenger mutations),對于起重要作用的驅(qū)動突變(driver mutations)的識別仍依賴于重復(fù)統(tǒng)計(jì)。蛋白質(zhì)組學(xué)技術(shù)主要基于液相色譜-串聯(lián)質(zhì)譜(LC-MS/MS),識別并量化在癌癥中被調(diào)控的蛋白質(zhì)和翻譯后修飾(PTMs),越來越多地用于癌癥研究。蛋白質(zhì)組學(xué)則直接提供了進(jìn)行中的蛋白質(zhì)調(diào)控和信號傳遞信息。更重要的是,蛋白質(zhì)組學(xué)還可對磷酸化、乙酰化和泛素化等PTMs深入分析,從而提供影響細(xì)胞信號、定位、分子復(fù)合體形成、翻譯和穩(wěn)定性失調(diào)的修飾信息,這些信息是無法通過DNA或mRNA分析反映的。
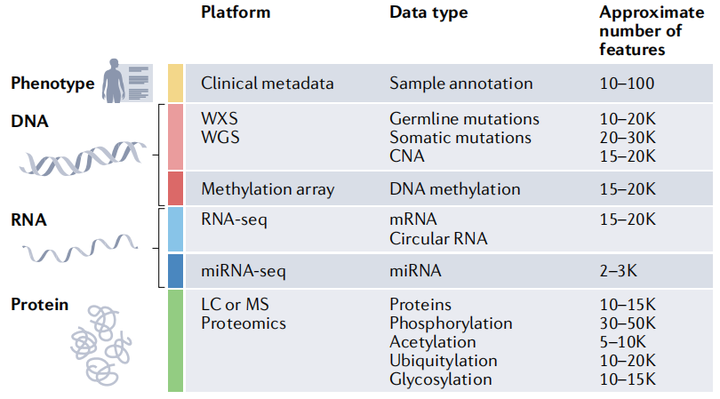
隨著乳腺癌、結(jié)腸癌和卵巢癌早期蛋白質(zhì)基因組學(xué)研究的發(fā)布,一系列針對不同類型腫瘤的蛋白質(zhì)基因組學(xué)圖譜的研究相繼發(fā)表。這些研究系統(tǒng)地整合了基因組、轉(zhuǎn)錄組、蛋白質(zhì)組和PTMs數(shù)據(jù),以進(jìn)一步了解疾病發(fā)病機(jī)制并確定每種癌癥的治療靶點(diǎn)。以下是已發(fā)表的蛋白質(zhì)基因組學(xué)研究對癌癥的深刻見解。
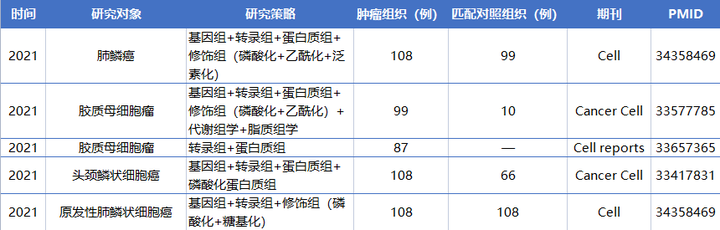
蛋白質(zhì)組數(shù)據(jù)比其他數(shù)據(jù)類型能夠更好地預(yù)測生存。對膠質(zhì)母細(xì)胞瘤的研究表明,蛋白質(zhì)組數(shù)據(jù)較RNA測序(RNA-seq)數(shù)據(jù)與患者生存的相關(guān)性更顯著。在一項(xiàng)前列腺癌研究中,蛋白質(zhì)組數(shù)據(jù)比任何其他個體數(shù)據(jù)類型更好地預(yù)測了患者復(fù)發(fā),它們與基因組或表觀基因組的結(jié)合進(jìn)一步提高了預(yù)測性能。免疫蛋白基因組分析顯示,在HNSCC腫瘤中存在廣泛的免疫細(xì)胞浸潤水平。這些腫瘤中多個免疫檢查點(diǎn)蛋白的一致性上調(diào)可能解釋了抗PD1單藥治療的中等反應(yīng)率,并為研究具有高水平免疫細(xì)胞浸潤(免疫h(yuǎn)ot)的腫瘤中的聯(lián)合檢查點(diǎn)封鎖提供了理論依據(jù)。
蛋白質(zhì)組學(xué)的加入提升了癌癥研究的廣度和深度。癌癥亞型主要是通過臨床、基因組或轉(zhuǎn)錄組特征來定義的。多組學(xué)方法可以根據(jù)基礎(chǔ)生物學(xué)和/或結(jié)局進(jìn)一步細(xì)化或重新定義亞型。在癌癥中,這種更精細(xì)的亞型劃分需求日益增長,有望被用于臨床水平,以定制治療策略和評估效果。目前的癌癥臨床實(shí)踐幾乎完全是由基因組學(xué)驅(qū)動的,早期研究報(bào)道,RNA水平和蛋白水平的平均相關(guān)性在0.3-0.45之間。多項(xiàng)研究探討了RNA和蛋白質(zhì)水平的相關(guān)性與穩(wěn)定性之間的關(guān)系,結(jié)果表明,在mRNA和蛋白質(zhì)中穩(wěn)定性相似的基因往往具有較高的RNA-蛋白質(zhì)相關(guān)性。此外,mRNA-蛋白質(zhì)相關(guān)性較低的基因往往是泛素蛋白酶體途徑的靶點(diǎn)或受miRNAs調(diào)控。因此,蛋白質(zhì)組學(xué)為癌癥研究增加了一個補(bǔ)充的維度。
02.蛋白質(zhì)基因組學(xué)研究中的樣品質(zhì)量要求
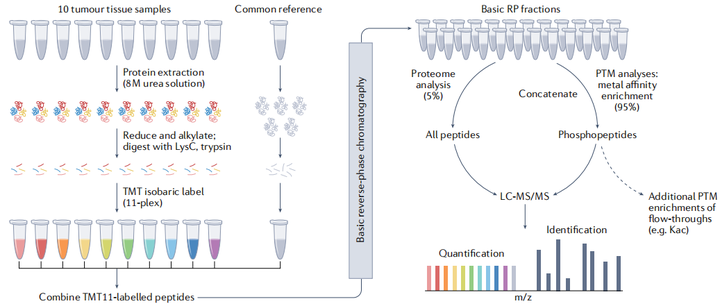
癌癥蛋白質(zhì)基因組學(xué)研究對樣本的采集和處理提出了特殊的要求,以保證數(shù)據(jù)質(zhì)量可靠并能真實(shí)反映潛在生命機(jī)理。雖然整體蛋白質(zhì)組在一定條件下是相對穩(wěn)定,但磷酸化等修飾蛋白質(zhì)組更具動態(tài)性,容易與組織缺血等其他損傷引起的效應(yīng)相混淆,經(jīng)過嚴(yán)格注釋的有條理的前瞻性收集可以最大限度地減少此類分析前變量并控制下游分析,因此需要在嚴(yán)格的SOP 下收集樣本,否則可能不適合 PTM分析。此外,制備技術(shù)如激光捕獲顯微解剖技術(shù)被用于分離腫瘤上皮細(xì)胞和其他感興趣的組分。
03.計(jì)算方法和工具簡述
結(jié)合來自不同隊(duì)列的多組學(xué)數(shù)據(jù)集進(jìn)行泛癌分析帶來了額外的挑戰(zhàn)。當(dāng)以計(jì)算方式組合這些數(shù)據(jù)集時需要校正數(shù)據(jù)集中的任何群組特定信號。整合蛋白質(zhì)基因組數(shù)據(jù)分析借鑒了統(tǒng)計(jì)學(xué)、機(jī)器學(xué)習(xí)和大數(shù)據(jù)分析等一系列學(xué)科的方法和算法。蛋白質(zhì)基因組數(shù)據(jù)分析方法分為三大類:以序列為中心的方法、蛋白質(zhì)基因組關(guān)系分析和綜合建模。

04.面向臨床應(yīng)用簡述
為了解決每個臨床假設(shè)并朝著臨床實(shí)用性邁進(jìn),從患者隊(duì)列中獲得處理一致的樣本并快速冷凍是研究的關(guān)鍵。新鮮冷凍(FF)、福爾馬林固定石蠟包埋(FFPE)和OCT包埋是腫瘤樣本的主要保存方法。單細(xì)胞蛋白質(zhì)組學(xué)允許從單細(xì)胞中分析大約1000個蛋白,隨著樣品處理、色譜和質(zhì)譜儀器的改進(jìn),單細(xì)胞蛋白質(zhì)組學(xué)技術(shù)的作用將進(jìn)一步發(fā)揮。從而和基因組數(shù)據(jù)整合以了解癌癥。
結(jié)論
隨著標(biāo)準(zhǔn)化、高通量的蛋白質(zhì)基因組學(xué)技術(shù)不斷發(fā)展,臨床研究將向著更大隊(duì)列的方向進(jìn)步,基因組學(xué)、表觀基因組學(xué)、轉(zhuǎn)錄組學(xué)、蛋白組學(xué)和翻譯后修飾組學(xué)等多組學(xué)數(shù)據(jù)的集成,已經(jīng)成為癌癥系統(tǒng)生物學(xué)的重要組成部分。
文獻(xiàn)下載鏈接:
https://pan.baidu.com/s/1ygc-Q2JiueS3geiJnRJfeA
提取碼:0000