帕金森小科普 - 概況
全球有大約 450 萬帕金森病患者,近一半在中國。中國目前已有 220 萬帕金森患者。我國 60 歲以上的老年人超過 1% 患有帕金森病,65 歲以上的老年人口中大約有 1.7% 的人患有帕金森病,70 歲以上患病率達(dá) 3%~5%,是繼腫瘤、心腦血管病之后中老年的 「第三殺手」,而且每年新發(fā)病例近十萬人。(來自網(wǎng)絡(luò))
帕金森小科普 - 發(fā)病機(jī)制及臨床表現(xiàn)
帕金森病是除阿爾茲海默癥之外的神經(jīng)退行性疾病。臨床上,該病的特征表現(xiàn)為動作遲緩、靜止性震顫及僵硬,這是由于中腦腹側(cè)黑質(zhì)部分缺乏多巴胺能神經(jīng)元所致(如下圖 1)。
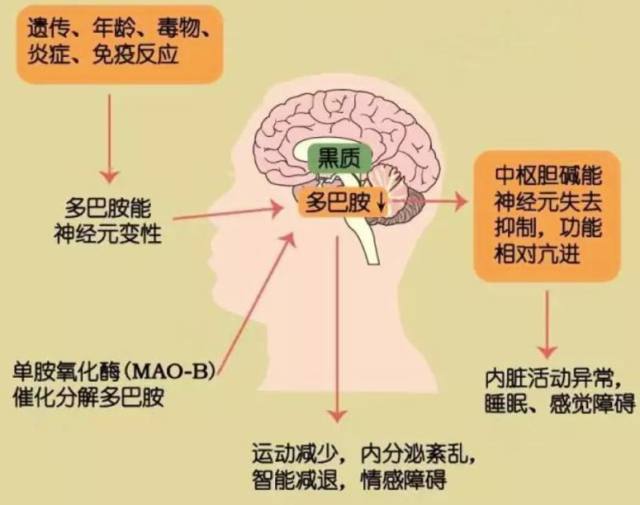
圖 1 帕金森發(fā)病機(jī)制及臨床表現(xiàn)
帕金森小科普 - 分子機(jī)制
正常情況下,突觸前神經(jīng)元中神經(jīng)遞質(zhì)多巴胺的釋放導(dǎo)致信號轉(zhuǎn)導(dǎo),通過 D1 和 D2 類型的多巴胺受體轉(zhuǎn)導(dǎo)至突觸后神經(jīng)元。D1 受體的信號轉(zhuǎn)導(dǎo)是通過 G 蛋白激活腺苷酸環(huán)化酶,形成環(huán)腺苷酸(cAMP)并激活 PKA 后完成的。D2 類型受體則通過抑制腺苷酸環(huán)化酶阻止此信號轉(zhuǎn)導(dǎo)。(1-2)(如圖 2, Normal State)。
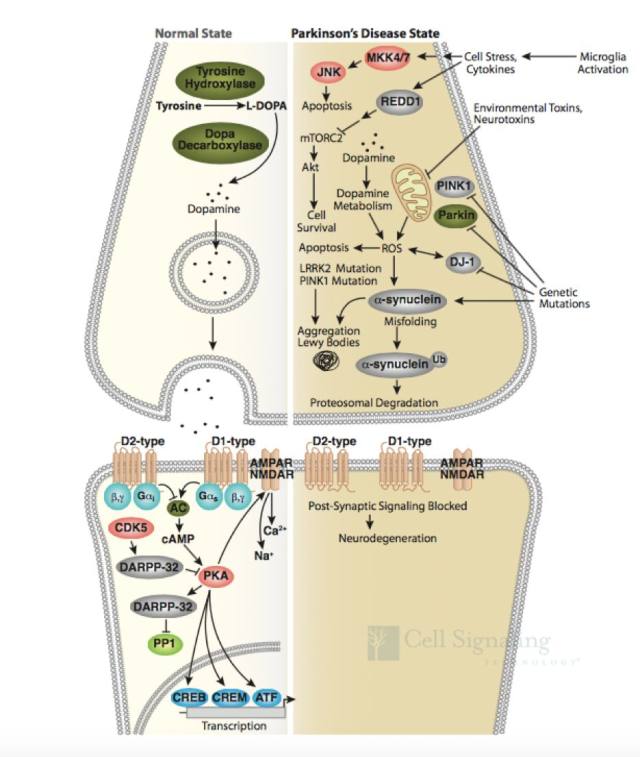
圖 2 帕金森病信號通路
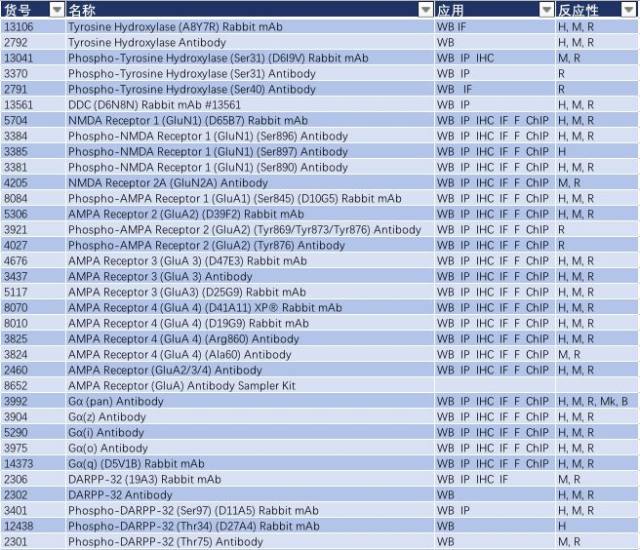
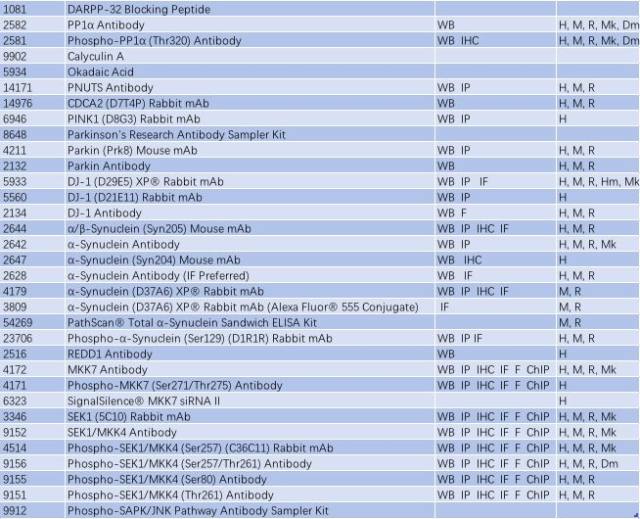
帕金森病可能是基因突變(家族性)和暴露于環(huán)境和神經(jīng)毒素下(散發(fā)性)導(dǎo)致。Parkin、DJ-1 和 PINK1 發(fā)生功能喪失性突變和隱性遺傳導(dǎo)致線粒體功能障礙和活性氧簇(ROS)積聚,而 α-突觸核蛋白(α-synuclein)和 LRRK2 的錯義突變和顯性遺傳可能影響蛋白降解通路,導(dǎo)致蛋白質(zhì)聚集以及路易小體積聚(如圖 2,Parkinsen’s Diease State),進(jìn)而導(dǎo)致多巴胺能神經(jīng)元過早退化或者導(dǎo)致多巴胺釋放和多巴胺能神經(jīng)傳遞受損,這可能是先于多巴胺能神經(jīng)元壞死的早期病理變化前兆(3-5)。因此,研究帕金森的發(fā)病分子機(jī)制,Parkin、DJ-1 、PINK1、α-synuclein 和 LRRK2 這些經(jīng)典的靶蛋白必不可少。CST 提供了這些靶蛋白的優(yōu)質(zhì)抗體,見表 1。
以上介紹是關(guān)于帕金森疾病的基礎(chǔ)知識,我們最想知道的 「信號通路」 將會逐漸登場。說到相關(guān)信號通路,我們還要再溫習(xí)一下帕金森病信號通路。從圖 2 我們看到,帕金森病還有一個炎癥部分,來源于小神經(jīng)膠質(zhì)細(xì)胞激活后導(dǎo)致炎癥細(xì)胞因子釋放和應(yīng)激。小神經(jīng)膠質(zhì)細(xì)胞激活可通過 JNK 通路導(dǎo)致細(xì)胞凋亡(這里的細(xì)胞凋亡大多是受 NF-κB 調(diào)控的),并通過 REDD1 阻止 Akt 信號轉(zhuǎn)導(dǎo)通路(如圖 2,Parkinsen’s Diease State)。
表 1:
與帕金森相關(guān)的信號通路 - NF-kB
研究顯示,炎癥和免疫反應(yīng)是帕金森疾病(PD)進(jìn)展的決定性因素,也是遺傳性和散發(fā)性 PD 發(fā)病的潛在因素之一。近來,研究報道的 PD marker 基因,如 SNCA (α-synuclein) 基因或 LRRK2(Leucine-rich repeat kinase 2), 通過激活小膠質(zhì)細(xì)胞和星形膠質(zhì)細(xì)胞刺激炎癥反應(yīng)直接參與 PD 的進(jìn)展(6)。還有文章報道早期的 PD 患者在大腦黑質(zhì)和硬核區(qū)存在激活的小膠質(zhì)細(xì)胞(7)。由炎癥信號通路的失調(diào)而引起小膠質(zhì)細(xì)胞的激活都涉及到 PD 的神經(jīng)炎癥反應(yīng)。激活的外周炎癥有助于 PD 的發(fā)生和(或)進(jìn)展,其與中樞炎癥反應(yīng)協(xié)同作用,促進(jìn)使黑質(zhì)致密部(SNpc)多巴胺能神經(jīng)元變性(8-9)。
NF-κB,自發(fā)現(xiàn)至今已有 30 多年,研究人員已經(jīng)證明 NF-κB 是各種促炎介質(zhì)基因表達(dá)的「總開關(guān)」(10),它在神經(jīng)系統(tǒng)的所有細(xì)胞中都表達(dá),包括神經(jīng)元、少突細(xì)胞、小膠質(zhì)細(xì)胞、星形膠質(zhì)細(xì)胞等(如圖 3)(11,12,16)。NF-κB 通過調(diào)控促炎細(xì)胞因子 tumor necrosis factor (TNF)、白介素 1 (Interleukin 1, IL-1)、趨化因子 monocyte chemoattractant protein(MCP-1)等在炎癥反應(yīng)中起到非常重要的作用(16)。臨床上用于應(yīng)對炎癥的氨基水楊酸鹽及脂多糖等藥物都是通過抑制 NF-κB 起作用的。同時,大量的研究表明,這些藥物可以抑制小膠質(zhì)細(xì)胞的激活,同時對多巴胺能神經(jīng)元有一定的保護(hù)作用(13)。盡管 NF-κB 激活可以防止自激活的細(xì)胞凋亡,但是作為一個轉(zhuǎn)錄因子,它可調(diào)控細(xì)胞毒性藥物如一氧化氮的產(chǎn)生,進(jìn)而可能間接導(dǎo)致其他細(xì)胞的凋亡(圖 3)。尤其是小膠質(zhì)細(xì)胞,在激活的時候會產(chǎn)生神經(jīng)毒性的活性氧等物質(zhì),這就解釋了細(xì)胞因子引起的小膠質(zhì)細(xì)胞激活是通過 NF-κB 激活(14)。脂多糖(LPS)能活化小膠質(zhì)細(xì)胞,進(jìn)而使 SNpc 部位多巴胺能神經(jīng)元退化(15)。
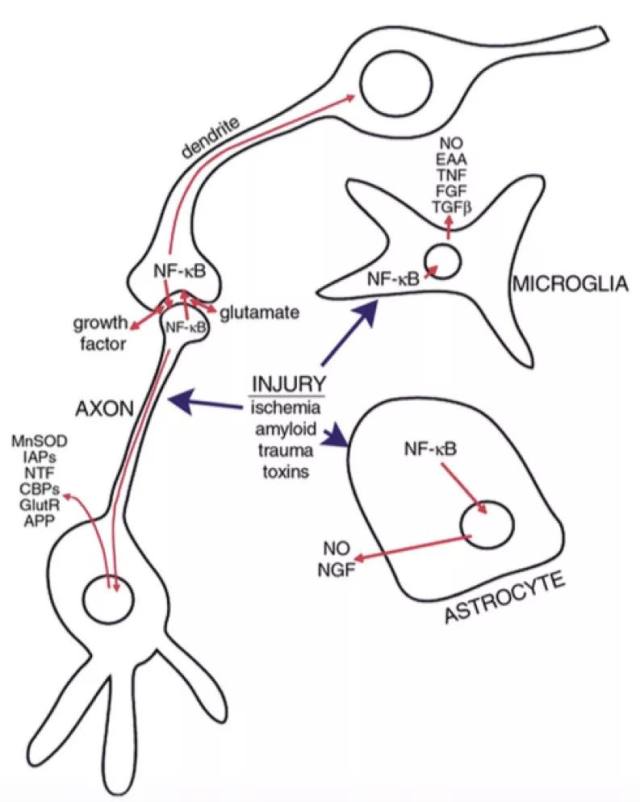
圖 3 激活的 NF-κB 在神經(jīng)元和膠質(zhì)細(xì)胞中的復(fù)雜作用。
腦損傷后,神經(jīng)元與膠質(zhì)細(xì)胞中的 NF-κB 被激活。NF-κB 激活的神經(jīng)元誘導(dǎo)調(diào)節(jié)突觸可塑性的基因表達(dá);NF-κB 激活的膠質(zhì)細(xì)胞產(chǎn)生促炎細(xì)胞因子和潛在的神經(jīng)毒性和刺激毒素(excitotoxins)(16)。
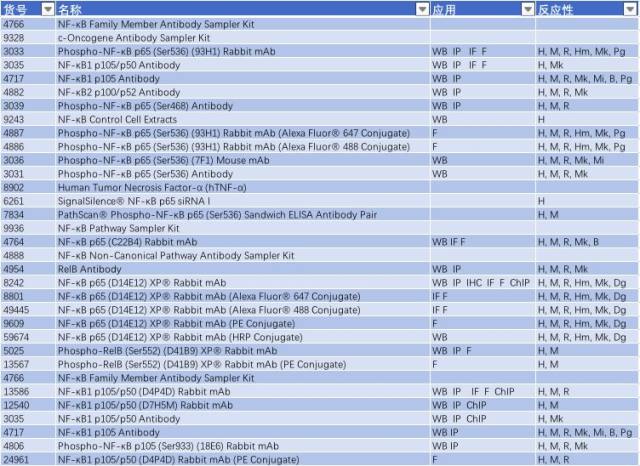
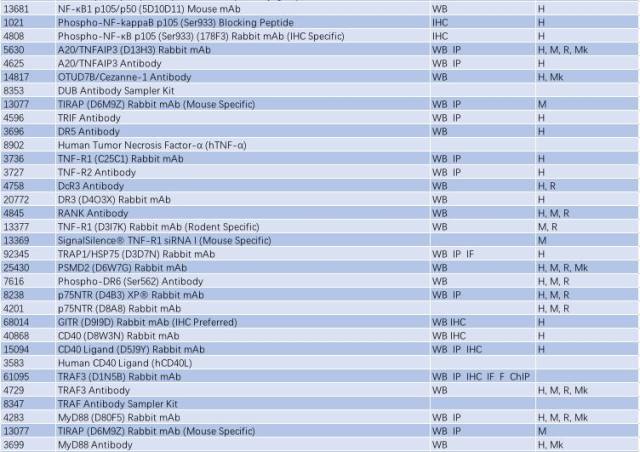
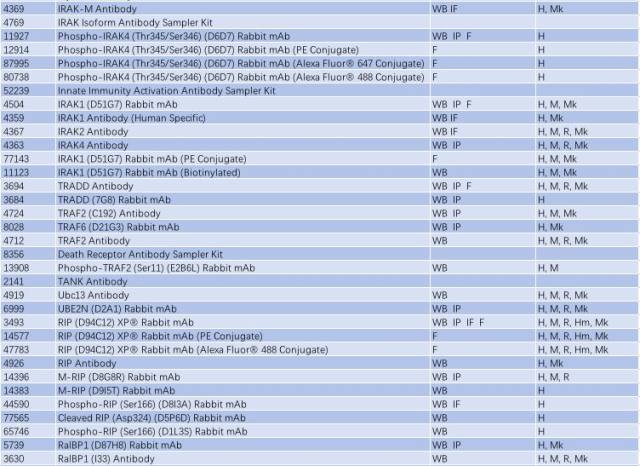
因此,NF-κB 信號通路的激活與慢性炎癥反應(yīng)密切相關(guān),抑制小神經(jīng)膠質(zhì)細(xì)胞中 NF-κB 的活性可能是有效治療 PD 的一個手段。研究 NF-κB 信號通路的激活可從以下靶點(diǎn)入手(表 2)。
表 2:
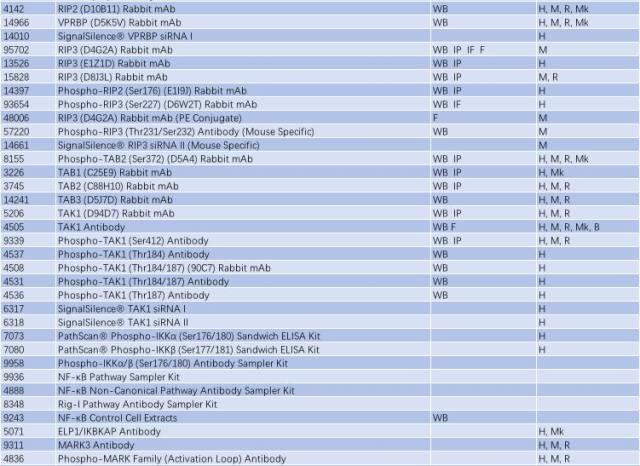
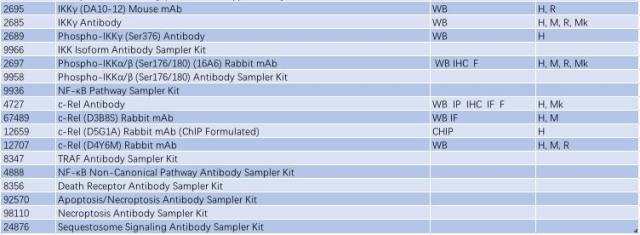
與帕金森相關(guān)的信號通路 - Jak/Stat
Jak/Stat 信號通路的激活對神經(jīng)炎癥及神經(jīng)退行性疾病的影響是近幾年中比較新的研究。Etty N.Benveniste 最近的研究報道了抑制 Jak/Stat 通路可以保護(hù)多巴胺神經(jīng)元的退化(17)。
Etty 為了證明激活的 Jak/Stat 信號通路與帕金森的關(guān)系,分別在細(xì)胞水平和動物模型上做了深入的研究,文章數(shù)據(jù)詳實(shí)、證據(jù)充分。通過 Jak1/2 抑制劑,AZD1480(該抑制劑可以減少 STAT1 和 STAT3 的激活)(如圖 4,圖中 P-STAT1 和 P-STAT3 的抗體都來自于 CST),抑制了小膠質(zhì)細(xì)胞和巨噬細(xì)胞中的 MHCII 和炎癥基因表達(dá)。在體內(nèi)的研究中,研究者們使用了由 α-synuclein 病毒過表達(dá)的大鼠 PD 模型。AZD1480 通過抑制小膠質(zhì)細(xì)胞活化、巨噬細(xì)胞和 CD4+ T - 細(xì)胞浸潤和促炎細(xì)胞因子/趨化因子的產(chǎn)生,抑制了 α-synuclein 誘導(dǎo)的神經(jīng)炎癥。大量參與細(xì)胞信號、神經(jīng)系統(tǒng)發(fā)育和功能、炎性疾病/過程,以及神經(jīng)系統(tǒng)疾病的基因,在 α-synuclein 過表達(dá)的大鼠黑質(zhì)中得到增強(qiáng),并在 AZD1480 處理后受到抑制(圖 5)。重要的是,體內(nèi)實(shí)驗還證明,抑制 JAK/Stat 通路的可抑制多巴胺能神經(jīng)元變性(圖 6)。
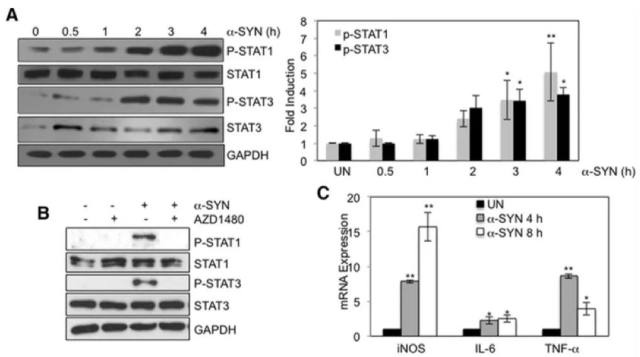
圖 4 α-SYN 對 Stat 的激活及基因表達(dá)的作用
注:A. 小鼠 BMDMs 被體外合成的α-SYN (500 nM) 刺激至 4 h 后,STAT1/3 及其磷酸化基因的表達(dá);B.BMDMs 在 AZD1480 (0.5 M) 的刺激 2 h 后,STAT1/3 磷酸化受到抑制;C. α-SYN 刺激引起炎癥相關(guān)基因的表達(dá)

圖 5 AZD1480 在體內(nèi)抑制了α-SYN 激活的 Jak/Stat 信號通路
A.VH 或者 AZD1480 (10 mg/kg/d) 灌胃 AAV2-GFP 或 AAV2-SYN 感染 2 周的大鼠,2 周后取出黑質(zhì)區(qū),進(jìn)行 RNA-seq;B. 聚類分析的熱圖,神經(jīng)炎癥等基因α-synuclein 過表達(dá)的大鼠黑質(zhì)中得到增強(qiáng),并在 AZD1480 處理后受到抑制。
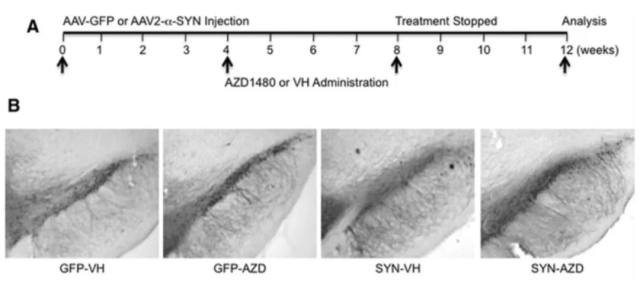
圖 6 AZD1480 對黑質(zhì)區(qū) TH 陽性神經(jīng)元的保護(hù)
A.AAV2- GFP 或 AAV2- SYN 感染大鼠 4 周和 VH 或者 AZD1480 (10 mg/kg/d) 處理進(jìn)程;B.α-SYN 3 周內(nèi)誘導(dǎo)了 50% 多巴胺能神經(jīng)的減少,AZD1480 的處理使得這種情況減少。
這里只是把其中的一小部分圖展示出來,若想看更詳細(xì)的內(nèi)容,請參考文章(17)。
此外,Piotr Przanowski 等報道了 Stat1 和 Stat3 誘導(dǎo)炎癥相關(guān)基因 Jmjd3 的表達(dá),以及促炎癥細(xì)胞因子的產(chǎn)生,從而激活小膠質(zhì)細(xì)胞。(18)。可見,Jak/Stat 信號通路也是通過一些神經(jīng)炎癥參與帕金森的進(jìn)程。更多相關(guān)知識請見:https://www.cst-c.com.cn/contents/science/cst-pathways/science-pathways 及產(chǎn)品請看表 3。
表 3:
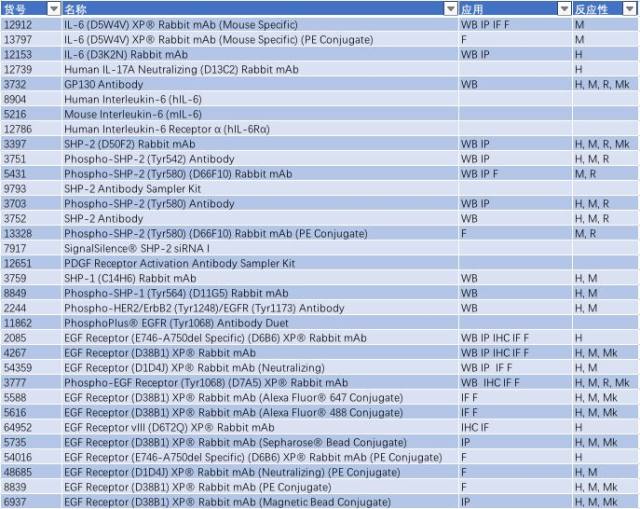
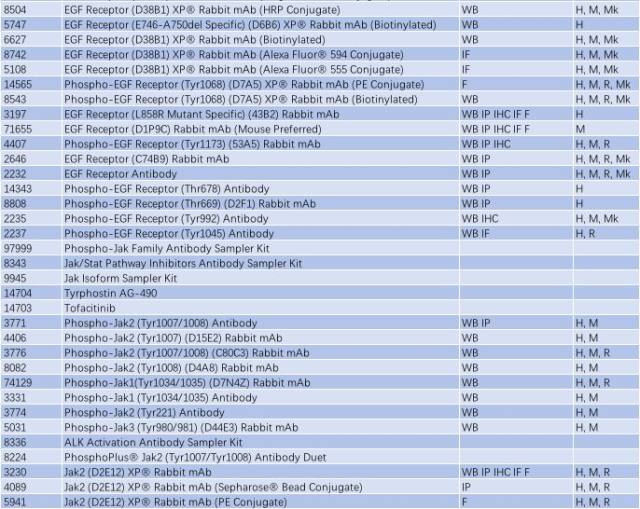
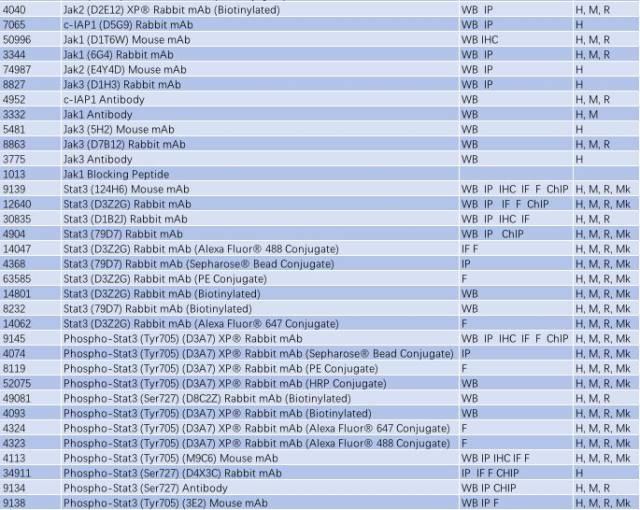

與帕金森相關(guān)的信號通路 - TLR
Toll 樣受體(TLRs)可識別不同的病原體相關(guān)分子模式,并在先天性免疫應(yīng)答中扮演著不可或缺的角色。它們是抵御病原體入侵的第一道防線,在炎癥、免疫細(xì)胞調(diào)控、存活和增殖方面發(fā)揮著關(guān)鍵作用。到目前為止,已發(fā)現(xiàn) 11 個 TLR 家族成員,其中 TLR1、TLR2、TLR4、TLR5、TLR6 和 TLR11 位于細(xì)胞表面,而 TLR3、TLR7、TLR8 和 TLR9 位于內(nèi)體/溶酶體部分。TLR 信號轉(zhuǎn)導(dǎo)通路的激活來源于細(xì)胞漿 Toll/IL-1 受體(TIR)的結(jié)構(gòu)域,該結(jié)構(gòu)域與 TIR 結(jié)構(gòu)域包含的接頭蛋白 MyD88 發(fā)生相互作用。經(jīng)過配體的刺激,通過兩個分子死亡結(jié)構(gòu)域的相互作用,MyD88 將 IL-1 受體相關(guān)激酶-4(IRAK-4)吸引到 TLRs。IRAK-1 經(jīng)磷酸化而激活并與 TRAF6 發(fā)生作用,從而激活 IKK 復(fù)合體并導(dǎo)致 NF-κB 的激活。激活的 NF-κB 被轉(zhuǎn)移到細(xì)胞核中,又會促進(jìn)各種各樣的促炎分子的表達(dá)。從活化的小膠質(zhì)細(xì)胞和局部微環(huán)境分泌的這些促炎分子增加了 SNpc 的氧化應(yīng)激,從而導(dǎo)致多巴胺能神經(jīng)元的退化(19-21)。
各種各樣的促炎和神經(jīng)毒性因子, 如過氧化物、TNF、IL 都是由激活的小膠質(zhì)細(xì)胞分泌的。同時,這些促炎性因子促進(jìn)小膠質(zhì)細(xì)胞產(chǎn)生 mcp-1,MIP-1,而這些蛋白也反過來影響神經(jīng)炎癥的發(fā)生,早期的細(xì)胞因子是通過與 Toll-like receptor (TLR) 4 結(jié)合而發(fā)揮作用。當(dāng)這些細(xì)胞因子釋放出來,進(jìn)而誘導(dǎo) Stat1 和 Stat3 的激活(22)。
可見,TLR 信號通路與 NF-κB 和 Jak/Stat 信號通路緊密連接參與到帕金森病的發(fā)生及發(fā)展。更多細(xì)節(jié),可以查看 TLR 信號通路。相關(guān)產(chǎn)品見表 4。
表 4:
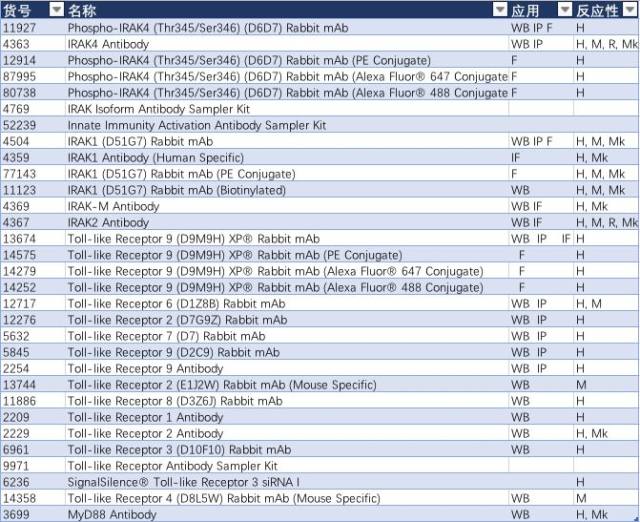
結(jié)語
總之,我們可以用一張圖來總結(jié)參與到帕金森病中的各神經(jīng)炎癥信號通路。
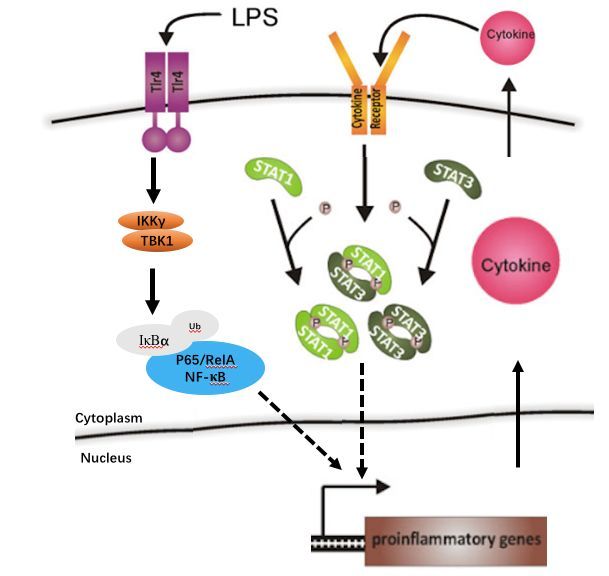
若您喜歡這篇文章,請快快收藏吧,說不定哪天就用上了。
參考文獻(xiàn):
1. Dauer W, Przedborski S.etal. Parkinson'sdisease: mechanisms and models. Neuron.2003;39(6), 889–909.
2. Girault JA, Greengard P.et al Theneurobiology of dopamine signaling. Arch Neurol. 2004;61(5), 641–4.
3. Imai Y, Lu B ,et al. Mitochondrialdynamics and mitophagy in Parkinson's disease: disordered cellular power plantbecomes a big deal in a major movement disorder. Curr Opin Neurobiol. 2011;21(6), 935–41.
4. Patten DA, Germain M,Kelly MA, Slack RS. Reactiveoxygen species: stuck in the middle of neurodegeneration. J. Alzheimers Dis. 2010;20 Suppl 2, S357–67.
5. Springer W, Kahle PJ .Regulationof PINK1-Parkin-mediated mitophagy. Autophagy.2011;7(3), 266–78.
6. Barcia C. Glial-mediated inflammation underlying parkinsonism. Scientifica (Cairo). 2013;357805.
7. Dzamko N, Geczy CL, Halliday GM. Inflammation isgenetically implicated in Parkinson’s disease. Neuroscience. 2015;302:89-102.
8. Bove J, Perier C. Neurotoxin-based models ofParkinson’s disease. Neuroscience. 2012;211:51-76.
9. Kanaan NM, Kordower JH, Collier TJ. Age-relatedchanges in glial cells of dopamine midbrain subregions in rhesus monkeys. Neurobiol Ag- ing. 2010;31(6):937-952.
10. TsoulfasG, GellerDA.NF-κBintransplantation:friendorfoe?Transpl Infect Dis. 2001;3(4):212-219.
11. Baeuerle, P., and Baltimore, D. NF-κB: ten yearsafter. Cell. 1996; 87:13–20.
12. O』Neill, L.A., and Kaltschmidt, C. NF-kappa B: acrucial tran- scription factor for glial and neuronal cell function. Trends Neurosci. 1997; 20:252–258.
13. Roebuck KA, Carpenter LR, Lakshminarayanan V,Page SM, Moy JN, Thomas LL. Stimulus-specific regulation of chemokineexpression involves differential activation of the redox-responsivetranscription factors AP-1 and NF-κB. JLeukoc Biol. 1999;65(3):291-298.
15. Liu M, Bing G.Lipopolysaccharide animal models for Parkinson’s disease. Parkinsons Dis. 2011;2011:327089.
16. NF-κB in neuronal plasticity and neurodegenerative disorders. Journal of ClinicalInvestigation, 2001;107(3), 247-254
17. Hongwei Qin,JessicaA. Buckley ,et al.Inhibition of the JAK/STAT PathwayProtects Against Synuclein-Induced Neuroinflammation and Dopaminergic . The Journal of Neuroscience, 2016 ;36(18):5144 –5159
18. Przanowski P, Dabrowski M, Ellert-Miklaszewska A, et al. Thesignal transducers Stat1 and Stat3 and their novel target Jmjd3 drive theexpression of inflammatory genes in microglia. J Mol Med (Berl). 2014;92(3):239- 254
19. Akira S, TakedaK. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499-511.
20. Banerjee A, Gerondakis S. Coordinating TLR-activated signalingpathways in cells of the immune system. Immunol Cell Biol. 2007;85(6):420-424.
21. Yamamoto M, Sato S, Hemmi H, et al. Role of adaptor TRIF in theMyD88-independent Toll-like receptor signaling pathway. Science. 2003;301(5633):640-643.
22. Tanaka S, IshiiA, Ohtaki H, Shioda S, Yoshida T, Numazawa S. Activa- tion of microglia inducessymptoms of Parkinson’s disease in wild-type, but not in IL-1 knockout mice. JNeuroinflammation. 2013;10:143
本文來自「CST博士互助平臺」,更多相關(guān)內(nèi)容,
請關(guān)注??

點(diǎn)擊此處,閱讀原文。