 期刊:Science China-Life Sciences
期刊:Science China-Life Sciences
影響因子:8.0
成簇的規(guī)則間隔短回文重復(fù)序列 (CRISPR)/Cas9 系統(tǒng)是編輯哺乳動物基因組的革命性方法。隨著慢病毒遞送方法的發(fā)展,CRISPR 篩選技術(shù)出現(xiàn),并實現(xiàn)了全基因組敲除的低成本方式。然而,CRISPR 篩選只能分析具有非常不同表型的基因,例如那些顯著影響細(xì)胞生長的基因,或者那些可以用抗體或熒光蛋白直接檢測到的基因。
2016 年,開發(fā)了一種稱為單細(xì)胞 CRISPR 篩選 (scCRISPR-seq) 的新技術(shù),該技術(shù)將 CRISPR 擾動和單細(xì)胞測序相結(jié)合,以實現(xiàn)大規(guī)模單細(xì)胞分辨率的混合遺傳篩選。scCRISPR-seq 的關(guān)鍵技術(shù)創(chuàng)新是慢病毒載體的創(chuàng)造性設(shè)計,稱為 Perturb-seq 載體,允許從測序中鑒定每個細(xì)胞中的 sgRNA(圖 16A)。scCRISPR-seq 可以促進(jìn)復(fù)雜調(diào)控機制和異質(zhì)細(xì)胞群的高通量功能解剖。
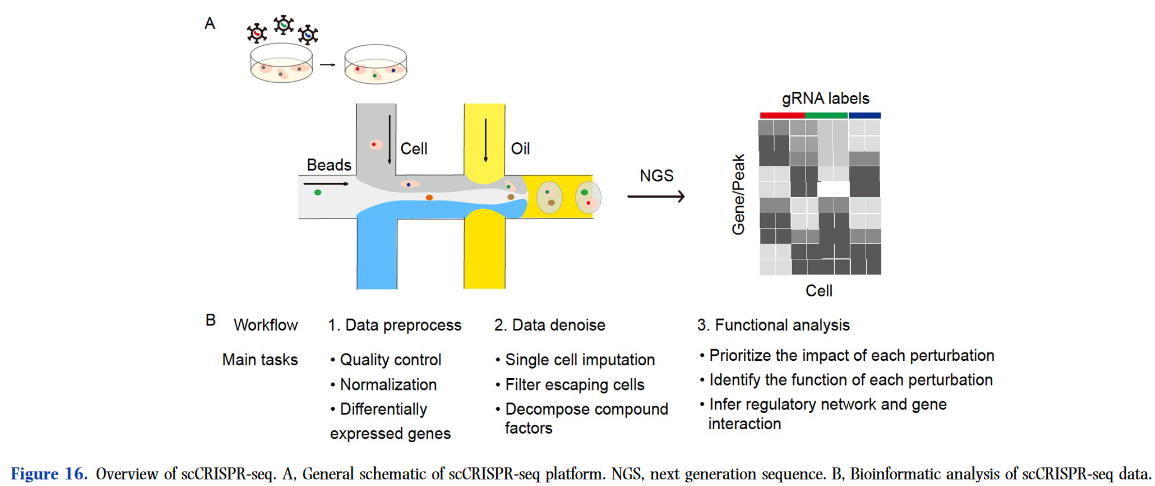
在本章中,我們將分四個不同的部分全面回顧 scCRISPR-seq。首先,我們將介紹 scCRISPR-seq 每個類別中的代表性技術(shù)。其次,我們將深入研究專門為分析 scCRISPR-seq 數(shù)據(jù)開發(fā)的主要工具。第三,我們將探索 scCRISPR-seq 的重要應(yīng)用。最后,我們將得出結(jié)論并參與與 scCRISPR-seq 相關(guān)的局限性和未來趨勢的討論。
scCRISPR-seq 平臺類別
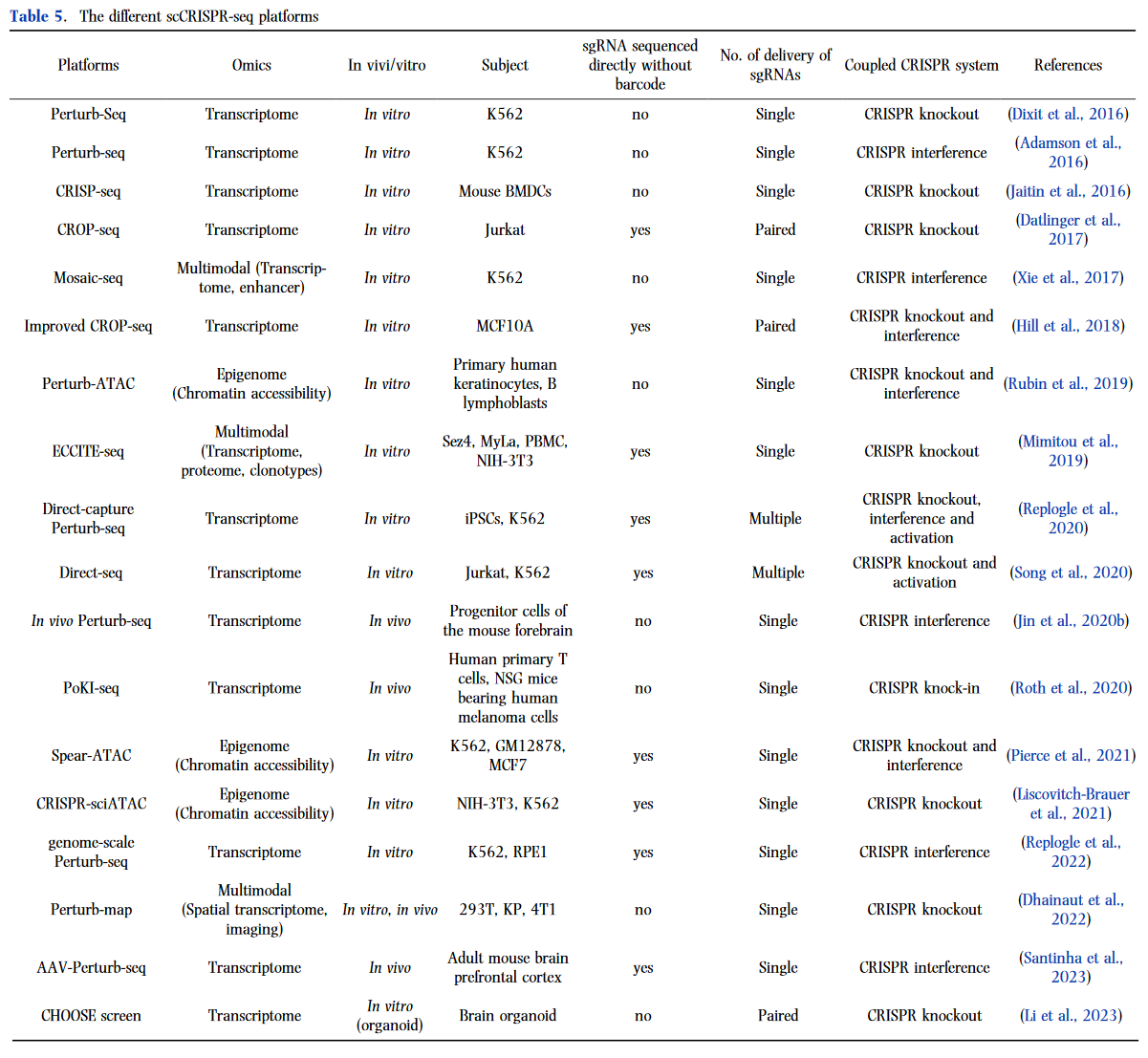
目前,已經(jīng)出現(xiàn)了許多替代的 scCRISPR-seq 平臺(表 5)。基于 scCRISPR-seq 的集成組學(xué)方法,這些平臺可分為三大類:基于轉(zhuǎn)錄組的 scCRISPR-seq、基于表觀基因組的 scCRISPR-seq 和多模態(tài) scCRISPR-seq。
基于轉(zhuǎn)錄組的 scCRISPR-seq
主要的 scCRISPR-seq 平臺是基于轉(zhuǎn)錄組的應(yīng)用程序,將 CRISPR 篩選與單細(xì)胞 RNA-seq 相結(jié)合。對于基于轉(zhuǎn)錄組的 scCRISPR-seq,Perturbseq 載體通常由單向?qū)?RNA (sgRNA)、細(xì)胞條形碼 (CBC)、基因條形碼 (GBC) 和 UMI 組成,例如 Perturb-seq和 CRISPseq。在 Perturb-seq 載體中,sgRNA 用于指導(dǎo) Cas9 核酸酶在目標(biāo)基因組區(qū)域誘導(dǎo)雙鏈斷裂,CBC 用于標(biāo)記每個細(xì)胞,而 GBC 用于標(biāo)記每個 sgRNA,UMI 用于標(biāo)記每個轉(zhuǎn)錄本。Perutrb-seq 和 CRISP-seq 是第一個開發(fā)的 scCRISPR-seq 平臺。這些方法涉及 Perturb-seq 載體的復(fù)雜構(gòu)建,包括復(fù)雜的克隆策略,有時還需要 gRNA 間隔區(qū)與其條形碼的解耦,這限制了它們的多功能性。CROPseq 優(yōu)化了 Perturbseq 載體的設(shè)計,通過向 Perturb-seq 載體添加 Poly-A 尾部來檢測每個細(xì)胞中誘導(dǎo)的 sgRNA 與 mRNA 偶聯(lián),這大大降低了 scCRISPR-seq 的復(fù)雜性和成本。然而,Hill 等人。(2018) 表明,由于這些研究的 Perturb-seq 載體設(shè)計,現(xiàn)有研究的慢病毒交換率僅為 50% 左右。因此,他們通過將向?qū)?RNA 作為條形碼來優(yōu)化 CROP-seq 載體設(shè)計,將交換率提高到 94%。由于 Perturb-seq 載體設(shè)計的限制,Perturb-seq 和 CRISP-seq 的每個慢病毒載體只能向細(xì)胞遞送單個編碼的 sgRNA,而 CROP-seq 能夠?qū)⒊蓪Φ?sgRNA 遞送到細(xì)胞。也就是說,它們都與多個 sgRNA 的遞送不相容。為了解決這個問題,Replogle 等人。(2020) 設(shè)計了直接捕獲 Perturb-seq,其中表達(dá)的 sgRNA 與單細(xì)胞轉(zhuǎn)錄組一起測序,并能夠遞送多個 sgRNA。直接捕獲 Perturbseq 對于遺傳相互作用的機制解剖特別有價值。它進(jìn)一步降低了 Perturb-seq 實驗的成本。Direct-seq 具有與直接捕獲 Perturb-seq 類似的功能,可實現(xiàn) CRISPR 擾動及其轉(zhuǎn)錄讀數(shù)一起分析,并支持多個 sgRNA 的遞送。2022 年,Replogle 等人引入了基因組規(guī)模的 Perturb-seq 方法。(2022 年),能夠?qū)τ绊?9,867 個基因的基因組規(guī)模遺傳擾動進(jìn)行公正和全面的分析。這一突破促進(jìn)了系統(tǒng)的基因功能分配和復(fù)雜細(xì)胞表型的探索。最近,利塔爾.(2023) 開發(fā)了 CRISPR-人類類器官-單細(xì)胞 RNA 測序 (CHOOSE) 系統(tǒng)。這種創(chuàng)新系統(tǒng)能夠進(jìn)行遺傳破壞和單細(xì)胞轉(zhuǎn)錄組學(xué),用于混合功能喪失篩選嵌合類器官。然而,上述所有 scCRISPR-seq 平臺都僅限于體外應(yīng)用。相比之下,體內(nèi)測定更具吸引力,因為它們與真實的有機條件更相似。因此開發(fā)了體內(nèi) Perturb-seq,這是 Perturb-seq 方案的一種變體,涉及在體內(nèi)進(jìn)行的合并擾動。此外,PoKI-seq (Roth et., 2020) 已經(jīng)證明了體內(nèi)研究重編程 T 細(xì)胞對實體瘤的免疫反應(yīng)的可行性。最近,Santinha 等人。(2023) 開發(fā)了腺相關(guān)病毒 (AAV) 介導(dǎo)的直接體內(nèi)單細(xì)胞 CRISPR 篩選,稱為 AAV-Perturb-seq,一種可用于體內(nèi)遺傳擾動轉(zhuǎn)錄連鎖分析和表型分析的可調(diào)且廣泛適用的方法。
基于表觀基因組的 scCRISPR-seq
基于表觀基因組的 scCRISPR-seq 除了轉(zhuǎn)錄組應(yīng)用外,還有基于表觀遺傳學(xué)的 scCRISPR-seq 平臺。2019 年開發(fā)了 Perturb-ATAC,這是一種將 CRISPR 干擾或敲除與單細(xì)胞染色質(zhì)可及性分析相結(jié)合的方法,該方法基于通過測序檢測轉(zhuǎn)座酶可及性染色質(zhì) (ATAC-seq) 同時檢測 CRISPR 向?qū)?RNA 和開放染色質(zhì)位點。他們應(yīng)用這種方法來確定各種反式調(diào)控因子的作用,包括 TFs、染色質(zhì)修飾劑以及人類和病毒 ncRNA,這可能有助于剖析順式調(diào)控元件和 ncRNA 轉(zhuǎn)錄本已被證明對基因表達(dá)有影響的基因座(Cho 等人,2018b;Engreitz 等人,2016 年;Rubin 等人,2019 年)。Perturb-ATAC 將 scCRISPR-seq 研究擴展到表觀基因組領(lǐng)域,使 scCRISPR-seq 更強大、應(yīng)用更廣泛。然而,Perturb-ATAC 受到高成本和低吞吐量的限制。為了應(yīng)對這一限制,開發(fā)了 Spear-ATAC(Pierce 等人,2021 年)以實現(xiàn)顯著更高的細(xì)胞通量和大幅降低成本,提供了更實用的替代方案。此外,CRISPR-sciATAC (Liscovitch-Braueret al., 2021) 顯示出與 Spear-ATAC 相似的細(xì)胞通量和成本。然而,它對染色質(zhì)可及性的細(xì)微變化表現(xiàn)出有限的敏感性。
多模態(tài) scCRISPR-seq
多模式單細(xì)胞檢測提供了異質(zhì)細(xì)胞群的高分辨率快照,但上述 scCRISPR-seq 平臺都僅限于一種模式,例如轉(zhuǎn)錄組或表觀基因組。因此,為了將該技術(shù)同時應(yīng)用于多組學(xué),開發(fā)了多模態(tài) scCRISPR-seq。Xie et al. (2017) 通過索引 CRISPR 測序 (Mosaic-seq) 開發(fā)了Mosaic單細(xì)胞分析,以擾亂增強子并共同測量每個細(xì)胞的轉(zhuǎn)錄組及其誘導(dǎo)的 sgRNA。Mosaic-seq 提供了一種新穎的工具,以基于 inper擾動的方式詢問非編碼基因的功能。此外,Mimitou 等人。(2019) 通過測序開發(fā)了擴展的 CRISPR 兼容轉(zhuǎn)錄組和表位細(xì)胞索引 (ECCITE-seq),它允許同時檢測來自每個細(xì)胞的轉(zhuǎn)錄組、蛋白質(zhì)、克隆型和 CRISPR 擾動。通過構(gòu)建 ECCITEseq 抗體的 a49 標(biāo)記面板來分析人外周血單核細(xì)胞 (PBMC),他們恢復(fù)了許多重要結(jié)果(Fanok等人,2018 年;Stoeckius 等人,2017 年),證明了 ECCITE-seq 結(jié)合免疫表型、克隆型和轉(zhuǎn)錄組信息的能力。空間轉(zhuǎn)錄組學(xué)能夠表征基因表達(dá)譜,同時保留有關(guān)空間組織背景的信息,這為生物學(xué)的不同領(lǐng)域提供了新的見解,例如神經(jīng)科學(xué)、發(fā)育生物學(xué)和癌癥研究(Moses 和 Pachter,2022)(參見下面的空間部分)。最近,開發(fā)了一種新的多模態(tài) scCRISPR-seq,稱為 Perturb-map (Dhainaut et., 2022),以通過成像和空間轉(zhuǎn)錄組學(xué)實現(xiàn) CRISPR 篩選原位的多模態(tài)表型分析。Perturb-map 基于蛋白質(zhì)條形碼 (Pro-Code) 系統(tǒng),該系統(tǒng)使用幾個線性表位的三重體組合來創(chuàng)建更高階的唯一條形碼集(Wroblewska 等人,2018 年)。這些獨特的條形碼可以標(biāo)記表達(dá)不同 CRISPR gRNA 的細(xì)胞。需要注意的是,Perturb-map 是唯一能夠?qū)崿F(xiàn)體內(nèi) CRISPR 篩選與空間轉(zhuǎn)錄組相結(jié)合的 scCRISPR-seq 平臺,特別適用于鑒定腫瘤組成、組織和免疫的遺傳決定因素。Perturb-map 基于蛋白質(zhì)條形碼 (Pro-Code) 系統(tǒng),該系統(tǒng)使用幾個線性表位的三重體組合來創(chuàng)建更高階的唯一條形碼集(Wroblewska 等人,2018 年)。這些獨特的條形碼可以標(biāo)記表達(dá)不同 CRISPR gRNA 的細(xì)胞。需要注意的是,Perturb-map 是唯一能夠?qū)崿F(xiàn)體內(nèi) CRISPR 篩選與空間轉(zhuǎn)錄組相結(jié)合的 scCRISPR-seq 平臺,特別適用于鑒定腫瘤組成、組織和免疫的遺傳決定因素。Dhainaut 等人。(2022) 將 Perturb-map 應(yīng)用于 TME 的研究。他們在肺癌小鼠模型中敲除 35 個基因,發(fā)現(xiàn)敲除 Tgfbr2 可以促進(jìn) TME 重塑和免疫排斥。
scCRISPR-seq 數(shù)據(jù)分析工具
scCRISPR-seq 數(shù)據(jù)包含豐富的擾動信息,這對于在單細(xì)胞水平上探索基因型和表型之間的關(guān)聯(lián)具有天然的優(yōu)勢。例如,通過將 Perturb-seq 應(yīng)用于 K562 細(xì)胞系,Adamson 等人。(2016) 表明,PERK 的擾動對未折疊蛋白質(zhì)反應(yīng)的影響比 ATF6 和 IRE1α 更大。Datlinger 等人 (2017) 在用 CROP-seq 激活 T 細(xì)胞受體 (TCR) 的條件下擾動了 Jurkat 細(xì)胞系中的 23 個轉(zhuǎn)錄因子,發(fā)現(xiàn) LCK 、 ZAP70 和 LAT 的敲除對 TCR 激活信號傳導(dǎo)有很強的負(fù)面影響。然而,由于其固有的噪聲,scCRISPR-seq 數(shù)據(jù)的分析是一項重大挑戰(zhàn)。因此,已經(jīng)開發(fā)了幾種生物信息學(xué)工具來幫助分析 scCRISPR-seq 數(shù)據(jù)(支持信息中的表 S12)。通常,這些 scCRISPR-seq 數(shù)據(jù)分析工具側(cè)重于三個部分(圖 16B):(i) 數(shù)據(jù)預(yù)處理,包括質(zhì)量控制、歸一化和差異表達(dá)基因檢測,例如 MIMOSCA 、MUSIC和 SCREE 。(ii) 數(shù)據(jù)去噪,包括單細(xì)胞插補、轉(zhuǎn)義細(xì)胞過濾和復(fù)合因子分解,例如 MUSIC、mixscape 和 SCREE 。(iii) 功能分析,包括確定每個擾動影響的優(yōu)先級,確定每個擾動的功能,推斷調(diào)節(jié)網(wǎng)絡(luò)和基因相互作用,例如 MUSIC、Normalisr、scMAGeCK、Pando和 GEARS。具體來說,LRICA是通過低秩矩陣分解來解碼數(shù)據(jù)的驅(qū)動信號/分量。MIMOSCA是一種計算sg RNA與每個基因之間關(guān)系的計算框架。LRICA 和 MIMOSCA 是作為原型開發(fā)的,沒有可執(zhí)行且用戶友好的實現(xiàn)。因此,Duan 等人。(2019) 開發(fā)了 MUSIC,這是一個通用的計算框架,用于通過主題建模評估每個擾動的影響,它最初出現(xiàn)在機器學(xué)習(xí)和自然語言處理社區(qū)或特定文檔集中的潛在主題發(fā)現(xiàn)中。MUSIC 將基因型表型與對大量噪聲的耐受性聯(lián)系起來,并從三個角度分析 scCRISPR-seq 數(shù)據(jù),即優(yōu)先考慮基因擾動效應(yīng)作為整體擾動效應(yīng)、非功能性主題特異性方式,以及量化不同擾動之間的相關(guān)性。scMAGeCK 也是分析 scCRISPR-seq 數(shù)據(jù)的框架,它是從 MAGeCK 擴展而來的。scMAGeCK 包括兩個模塊,scMAGeCK-RRA 和 scMAGECK-LR,其中 scMAGeCK-RRA 用于通過負(fù)二項分布識別顯著富集的 sgRNA,scMAGeCK-LR 用于通過線性回歸評估受影響的基因。scMAGeCK 顯示出對假陽性的良好控制,并且比其他方法具有更好的靈敏度。除了 scCRISPR-seq 的通用計算框架外,一些工具還專注于數(shù)據(jù)去噪。例如,SCEPTRE 是為使用條件隨機化測試進(jìn)行 scCRISPR-seq 數(shù)據(jù)校準(zhǔn)而開發(fā)的。SCEPTRE 對 scCRISPR-seq 數(shù)據(jù)表現(xiàn)出良好的校準(zhǔn)和靈敏度,產(chǎn)生了數(shù)百種由正交生物學(xué)證據(jù)支持的新調(diào)控關(guān)系。Mixscape 旨在通過混合判別分析過濾逃逸的細(xì)胞 (細(xì)胞誘導(dǎo)的 sgRNA,但不表現(xiàn)出擾動效應(yīng)) 來提高 scCRISPR-seq 數(shù)據(jù)的信噪比。Normalisr 用于重建 scCRISPR-seq 數(shù)據(jù)的基因調(diào)控網(wǎng)絡(luò)。Wang 等人.(2022g) 強調(diào)了識別克隆細(xì)胞的重要性,因為它們可能導(dǎo)致 scCRISPR-seq 數(shù)據(jù)出現(xiàn)假陽性。SCREE 是 scCRISPR-seq 數(shù)據(jù)分析的綜合管道。與前面提到的最初專注于 scCRISPR-seq 中的數(shù)據(jù)去噪和挖掘的方法相比,GEARS 專門設(shè)計用于預(yù)測對單基因和多基因擾動的轉(zhuǎn)錄反應(yīng)。這些方法大大增強了 scCRISPR-seq 數(shù)據(jù)的分析。
scCRISPR-seq 的應(yīng)用
scCRISPR-seq 因其強大的能力而被廣泛應(yīng)用于各個領(lǐng)域,包括連接基因型與表型、剖析遺傳調(diào)控以及研究腫瘤和自閉癥等非特異性疾病的遺傳機制。
連接基因型和表型
與傳統(tǒng)的 CRISPR 篩選只能識別具有非常不同表型的基因相比,scCRISPR-seq 具有揭示任何基因功能的能力。因此,scCRISPR-seq 天然適合大規(guī)模地將基因型與表型聯(lián)系起來。例如,Jaitin 等。(2016) 揭示了 22 個 TFs 對 CRISP-seq 刺激的脂多糖 (LPS) 刺激的出生骨髓細(xì)胞 (BMC) 的抗病毒、炎癥或發(fā)育過程的調(diào)節(jié)作用。Adamson 等人。(2016) 通過 Perturb-seq 系統(tǒng)分析了 K562 細(xì)胞中 83 個未折疊蛋白反應(yīng) (UPR) 相關(guān)基因的影響。此外,基因組規(guī)模的 Perturb-seq(Replogle等人,2022 年)提供了對遺傳擾動(9,867 個基因)的公正、全面的分析,有助于系統(tǒng)剖析與基因翻譯和核糖體生物發(fā)生相關(guān)的基因之間的關(guān)系。
剖析遺傳調(diào)控
scCRISPR-seq 還用于剖析基因組元件之間的復(fù)雜關(guān)系,包括編碼基因、轉(zhuǎn)錄因子、染色質(zhì)調(diào)節(jié)因子、增強子和其他非編碼元件。例如,Adamson 等人。(2016) 使用 Perturb-seq 發(fā)現(xiàn)了三個 UPR 傳感器基因(ATF6、PERK 和 IRE1)之間的串?dāng)_。CROP-seq 擾亂了 LPS 刺激后 Jurkat 細(xì)胞中調(diào)節(jié) TCR 激活的 TFs,并揭示了 TFs 之間的關(guān)系。此外,用于增強子擾動的 scCRISPR-seq,例如 Mosaic-seq (Xie et tal., 2017),可以發(fā)現(xiàn)新的增強子-基因?qū)Α4送猓瑂cCRISPR-seq 與 scATAC-seq 偶聯(lián),如 Perturb-ATAC、Spear-ATAC 和 CRISPR-sciATAC 可以揭示人淋巴細(xì)胞和白血病細(xì)胞中的表觀遺傳景觀重塑劑
研究遺傳機制
有幾種體內(nèi) scCRISPR-seq 平臺可用,能夠研究腫瘤和自閉癥等非特異性疾病的遺傳機制。例如,Perturb-map (Dhainaut et., 2022) 有助于識別與腫瘤組成、組織和免疫相關(guān)的遺傳決定因素。Using Perturbmap, Dhainaut etal.(2022) 發(fā)現(xiàn) TGFBR2 肺癌細(xì)胞的敲除促進(jìn)了腫瘤微環(huán)境重塑和免疫排斥。Roth 等人。(2020) 使用 PoKI-seq 對增強 T 細(xì)胞抗腫瘤功能的嵌合抗原受體進(jìn)行了篩選,提高了免疫抑制條件下的腫瘤浸潤和細(xì)胞殺傷率 內(nèi)黑色素瘤。此外,Jin 等人。(2020b) 使用 invivo Perturb-seq 評估了 35 個與自閉癥譜系障礙/神經(jīng)發(fā)育遲緩 (ASD/ND) 相關(guān)的 denovo 功能喪失風(fēng)險基因。他們從神經(jīng)元和神經(jīng)膠質(zhì)細(xì)胞類別中鑒定了細(xì)胞類型特異性和進(jìn)化上保守的基因模塊。利塔爾。(2023) 還關(guān)注了這些高危自閉癥譜系障礙基因,他們揭示了它們對使用 CHOOSE 系統(tǒng)在嵌花類器官中決定細(xì)胞命運的影響。最近,Santinha 等人。(2023) 使用 AAV-Perturb-seq 系統(tǒng)分析了與成年小鼠大腦前額葉皮層中 22q11.2 缺失綜合征基因相關(guān)的表型景觀。他們確定了三個 22q11.2 連鎖基因,這些基因積極參與已建立的和以前未識別的體內(nèi)神經(jīng)元功能控制途徑。
總結(jié)
在本章中,我們對 scCRISPR-seq 進(jìn)行了全面的回顧,分為三個不同的部分,其中包括 scCRISPR-seq 的類別、scCRISPR-seq 數(shù)據(jù)分析的工具以及 scCRISPR-seq 的顯著應(yīng)用。scCRISPR-seq 一直是功能基因組學(xué)研究的強大方法 (Bock et al., 2022)。在本節(jié)中,我們根據(jù)其綜合組學(xué)方法將 scCRISPR-seq 分為三個主要類別:基于轉(zhuǎn)錄組的 scCRISPRseq、基于表觀基因組的 scCRISPR-seq 和多模態(tài) scCRISPRseq。鑒于 scCRISPR-seq 數(shù)據(jù)中固有的噪聲,已經(jīng)開發(fā)了多種生物信息學(xué)工具來幫助初始化分析,從而沒有顯著改進(jìn)。
scCRISPR-seq 的多功能性使其在各個領(lǐng)域得到廣泛應(yīng)用,提供了強大的功能,例如連接基因型與表型、剖析遺傳調(diào)控以及探索腫瘤和自閉癥等特定疾病的遺傳機制。然而,在生物學(xué)研究中更廣泛地采用之前,需要注意三個關(guān)鍵方面:(i) 降低復(fù)雜性和成本:應(yīng)努力進(jìn)一步簡化和降低 scCRISPR-seq 實驗的復(fù)雜性和成本。這將增強可擴展性和可及性,使更多實驗室能夠利用這項技術(shù)。(ii) 擴大對復(fù)雜組織和體內(nèi)設(shè)置的適用性:雖然目前的 scCRISPR-seq 平臺主要針對細(xì)胞系,但迫切需要開發(fā)更強大的 scCRISPR-seq 平臺,這些平臺可以應(yīng)用于更復(fù)雜的組織,包括類器官,以及理想的體內(nèi)設(shè)置。這種擴展將使更廣泛的生物學(xué)研究成為可能。(iii) 降噪技術(shù):隨著 scCRISPR-seq 平臺數(shù)量的增加,開發(fā)更強大的方法來破譯 scCRISPR-seq 數(shù)據(jù)中的固有噪聲變得至關(guān)重要。這些方法將有助于 scCRISPR-seq 結(jié)果的可靠性和可解釋性,進(jìn)一步提高它們在不同研究環(huán)境中的實用性。
結(jié)尾
scRNA-seq 技術(shù)引起了世界各地許多科學(xué)家的廣泛關(guān)注,因為它具有在單細(xì)胞水平上研究細(xì)胞異質(zhì)性的優(yōu)勢。自新時代 inscRNA-seq 研究成立以來,已經(jīng)過去了僅僅 14 年,在此之前,Tang 等人取得了初步的概念和技術(shù)突破。(2009) 在 2009 年。在測序技術(shù)和生物信息學(xué)不斷發(fā)展的推動下,scRNA-seq 研究領(lǐng)域目前正在經(jīng)歷研究的激增。scRNA-seq 技術(shù)的成熟極大地促進(jìn)了其他單細(xì)胞組學(xué)研究的進(jìn)步。目前,單細(xì)胞組學(xué)檢測已擴展到基因組 (Dey et al., 2015)、表觀基因組 (Muto et al., 2021)、空間轉(zhuǎn)錄組學(xué) (Chen et al., 2022)、蛋白質(zhì)組學(xué) (Petersonet al., 2017;Specht et al., 2021)、代謝組學(xué) (Shrestha, 2020) 等多組學(xué)水平 (Angermueller et al., 2016),為單細(xì)胞水平研究提供更全面、更精細(xì)、更完整的分析策略。在這篇綜述中,我們總結(jié)了單細(xì)胞組學(xué)技術(shù)、數(shù)據(jù)分析及其應(yīng)用的最新進(jìn)展,概述了單細(xì)胞測序領(lǐng)域跨多個層次的前景。
在第 1 章中,我們?nèi)娓攀隽四壳翱捎玫?scRNA-seq 技術(shù)、實驗方法、數(shù)據(jù)分析程序及其在生物醫(yī)學(xué)領(lǐng)域的應(yīng)用。最初,通過分離單細(xì)胞并獨立構(gòu)建測序文庫來進(jìn)行單細(xì)胞測序。這些單細(xì)胞測序技術(shù)只能檢測少量細(xì)胞(數(shù)到數(shù)百個),例如 Tang 方法、STRT-seq 和 SMART-seq(Islam 等人,2012 年;Ramsköld 等人,2012 年;Tang 等人,2009 年)。然而,隨著對測序技術(shù)的深入研究,基于條形碼標(biāo)簽的單細(xì)胞鑒定已經(jīng)出現(xiàn),以及基于微滴或微孔的新型單細(xì)胞分離技術(shù)的出現(xiàn),如 Drop-Seq 和 Cyto-Seq (Fan et., 2015a;Macosko 等人,2015 年),單細(xì)胞轉(zhuǎn)錄組測序已進(jìn)入高通量時代。測序成本顯著降低,同時自動化和通量顯著提高。ScRNAseq 技術(shù)解決了細(xì)胞異質(zhì)性問題,為臨床疾病尤其是腫瘤的個體化治療開辟了新途徑,促進(jìn)了精準(zhǔn)醫(yī)療的發(fā)展。然而,由于起始材料量少,scRNA-seq 存在 oflow 捕獲效率和高脫落率的局限性。與大量 RNA-seq 相比,scRNA-seq 產(chǎn)生的數(shù)據(jù)噪聲更大且可變性更強。盡管研究人員已經(jīng)設(shè)計了多種工具來進(jìn)行不同的 scRNA-seq 數(shù)據(jù)分析,但技術(shù)噪聲和生物變異(例如,隨機轉(zhuǎn)錄)仍然對 scRNA-seq 數(shù)據(jù)的計算分析構(gòu)成巨大挑戰(zhàn)(Chen 等人,2019a)。因此,數(shù)據(jù)分析方法仍需進(jìn)一步優(yōu)化和改進(jìn)。
與日益成熟的 scRNA-seq 技術(shù)相比,其他單細(xì)胞組學(xué)技術(shù)仍處于萌芽階段。在第 2、3、4 和 5 章中,我們重點介紹了過去十年中單細(xì)胞基因組、表觀基因組、蛋白質(zhì)組學(xué)和代謝組學(xué)測序的最新工具、計算方法和應(yīng)用。ScWGS 徹底改變了我們對遺傳變異及其對人類健康和疾病影響的理解。它的快速發(fā)展加速了基因組研究,實現(xiàn)了個性化醫(yī)療,并為疾病的遺傳基礎(chǔ)和人類基因組多樣性提供了有價值的見解。細(xì)胞在染色質(zhì)可及性、核小體定位、組蛋白修飾和 DNA 甲基化方面表現(xiàn)出廣泛的異質(zhì)性。在單細(xì)胞樣品中繪制這些表觀基因組信息對于發(fā)育生物學(xué)、癌癥研究以及很快的發(fā)展非常重要。單細(xì)胞表觀基因組測序方法的進(jìn)步使單細(xì)胞染色質(zhì)狀態(tài)的高分辨率圖譜成為可能。然而,如今,單細(xì)胞表觀基因組技術(shù)存在數(shù)據(jù)丟失的問題。因此,盡管單個細(xì)胞表觀基因組數(shù)據(jù)集是聚類分析和基于大量目標(biāo)位點集合揭示細(xì)胞異質(zhì)性的強大資源,但它們提供單個目標(biāo)位點信息的能力非常有限(Carter 和 Zhao,2021)。因此,在未來的研究中,需要提高各種單個細(xì)胞表觀基因組測定中染色質(zhì)靶位點的覆蓋率,這將有助于理解整個細(xì)胞水平和單個特異性位點的細(xì)胞異質(zhì)性。單細(xì)胞蛋白質(zhì)組學(xué)由于其成分復(fù)雜、豐度低、動態(tài)范圍寬、缺乏擴增能力,處于爆發(fā)性發(fā)展的早期階段。就在 2019 年,對單細(xì)胞蛋白質(zhì)組的分析被描述為“夢想”,但今天已經(jīng)開發(fā)了幾種有前途的工具(Marx,2019)。我們相信,隨著可及性的優(yōu)化和通量的進(jìn)一步提高,單細(xì)胞蛋白質(zhì)組學(xué)在科學(xué)和臨床研究中真正的大規(guī)模應(yīng)用,如器官圖譜、藥物篩選和精確的疾病分類,是觸手可及的。單細(xì)胞代謝組學(xué)用于鑒定單細(xì)胞中代謝物的組成,測量其豐度,并研究其動態(tài)變化。同時,代謝組代表基因組、轉(zhuǎn)錄組和蛋白質(zhì)組的下游產(chǎn)物,并提供了功能的更直接和動態(tài)快照(Shrestha,2020 年)。總體而言,單細(xì)胞組學(xué)技術(shù)仍處于萌芽階段,它們將繼續(xù)蓬勃發(fā)展。
單個細(xì)胞是生命的基本單位。對單個細(xì)胞進(jìn)行多組學(xué)分析可以深入了解細(xì)胞的表型、疾病狀態(tài)和環(huán)境影響。在第 6 章、第 7 章和第 8 章中,我們?nèi)婵偨Y(jié)了多組學(xué)的綜合分析、scRNA-seq 和 CRISPR 篩選的聯(lián)合應(yīng)用以及空間轉(zhuǎn)錄組。在復(fù)雜的生物過程中,例如腫瘤發(fā)生和衰老,異質(zhì)性發(fā)生在不同的層面,包括基因組、轉(zhuǎn)錄組、蛋白質(zhì)組和表觀基因組。如果僅從單個細(xì)胞 atatime 分析一個成分,則只能檢測到基因調(diào)控網(wǎng)絡(luò)的局部概況,而無法準(zhǔn)確預(yù)測復(fù)雜的全球情況。在這種情況下,多組學(xué)技術(shù)凸顯其獨特的優(yōu)勢,可以在復(fù)雜組織的研究中提供更完整的基因調(diào)控網(wǎng)絡(luò)圖譜。對于空間轉(zhuǎn)錄組學(xué),它能夠在保留空間信息的情況下測量基因表達(dá),這將有利于研究細(xì)胞間關(guān)系和發(fā)現(xiàn)空間背景下的新調(diào)控機制。此外,空間轉(zhuǎn)錄組學(xué)使探索細(xì)胞命運決定的空間調(diào)控機制和組織模式的結(jié)構(gòu)成為可能。與傳統(tǒng)的 CRISPR 雜交篩選相比,scRNA-seq 和 CRISPR 的組合不僅可以在單個實驗中篩選數(shù)千個 gRNA,還可以同時捕獲擾動的全轉(zhuǎn)錄組數(shù)據(jù),以最清楚地了解細(xì)胞類型特異性基因功能和通路分析。因此,這些技術(shù)的結(jié)合可以更好、更深入地理解關(guān)鍵的生物過程和機制,這是未來單細(xì)胞技術(shù)發(fā)展的重要方向。
如今,單細(xì)胞組學(xué)技術(shù)在通量和分辨率方面都取得了重大進(jìn)步。展望未來,單細(xì)胞技術(shù)發(fā)展的主要趨勢是提高單細(xì)胞分選的效率和通量,增強測序覆蓋率和靈敏度,實現(xiàn)高通量多組學(xué)研究,并開發(fā)更多自動化的單細(xì)胞技術(shù)平臺,這將有助于降低單細(xì)胞技術(shù)的成本和技術(shù)門檻。單細(xì)胞技術(shù)有望在科學(xué)研究和研究轉(zhuǎn)化領(lǐng)域得到廣泛應(yīng)用,并將對健康監(jiān)測、疾病診斷和治療做出巨大貢獻(xiàn)。
參考文獻(xiàn):
Sun F, Li H, Sun D, et al. Single-cell omics: experimental workflow, data analyses and applications. Sci China Life Sci. 2025;68(1):5-102. doi:10.1007/s11427-023-2561-0